






JMLS 2023 December;8(2):93-103. 10.23005/ksmls.2023.8.2.93 Epub 2023 December 16
Copyright © 2023 by The Korean Society of Marine Life Science
Application of Environmental DNA (eDNA) for Marine Biodiversity Analysis
Soyun Choi; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Seung Jae Lee; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Eunkyung Choi; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Euna Jo; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Jinmu Kim; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Minjoo Cho; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Jangyeon Kim; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Sooyeon Kwon; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Hyun Park; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
- Abstract
eDNA (environmental DNA)란 특정 환경에 서식하는 생물로부터 유래한 DNA를 의미한다. 환경 시료로부터 추출한 eDNA를 활용하면 해당 환경에 서식하는 생물들의 효율적이고 정확한 모니터링을 수행할 수 있다. 해수 시료로부터 얻은 eDNA를 기반으로 해양생물 다양성 연구를 수행할 수 있다. 해수 시료를 채집하고 이로부터 eDNA를 추출한 뒤, metagenome 분석을 통해 서식하는 해양생물의 종 동정과 다양성 분석이 가능하다. 본 리뷰에서는 이처럼 해수의 eDNA를 활용하여 해양 지역의 생물 다양성 연구를 수행하는 전체적인 과정을 제시하고 있다. 아직 국내에는 해양생물 다양성 연구를 위해 eDNA를 적용하는 방법이 보편화 되어있지 않으며, 본 리뷰를 기반으로 이와 같은 eDNA 연구 방법을 정립하는데 도움을 줄 수 있을 것이다.
eDNA, an abbreviation for environmental DNA, means DNA derived from organisms inhabiting in a specific environment. The utilization of eDNA extracted from environmental samples allows for efficient and accurate monitoring of organisms inhabiting the respective environment. Specifically, eDNA obtained from seawater samples can be used to analyze marine biodiversity. After collecting seawater samples and extracting eDNA, metagenome analysis enables the taxonomic and diversity analysis among marine organisms inhabiting the sampled area. This review proposed an overall process of marine biodiversity analysis by utilizing eDNA from seawater. Currently, the application of eDNA for analyzing marine biodiversity in domestic setting is not yet widespread. This review can contribute to establishment of marine eDNA research methods in Korea, providing valuable assistance in standardizing the use of eDNA in marine biodiversity studies.
Keywords: eDNA(환경유전자) Metagenome(군 유전체) NGS(차세대 염기서열 분석) Marine organisms(해양생물) Biodiversity(생물다양성)
Correspondence to: Hyun Park; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
- Received
- 28 August 2023;
- Revised
- 25 September 2023;
- Accepted
- 11 October 2023.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Language: Korean/English,
Full Text:

1. 서론
환경유전자, 즉 eDNA란 environmental DNA의 약자로 수중이나 토양 및 대기와 같은 특정 환경에 존재하는 생물체에서 유래한 DNA를 의미한다(Taberlet et al., 2012; Wilcox et al., 2013). 생물들은 서식하고 있는 환경에서 배설물이나 각질, 점액 등을 체외로 분비하는데, 이러한 DNA 분자들이 모여 eDNA를 구성한다(Barnes et al., 2014). 환경 시료로부터 얻은 eDNA를 활용하면 해당 환경에 어떤 생물이 서식하는지 간접적으로 확인하고, 더 나아가 생물 종의 구성과 분포 및 다양성과 관련된 다양한 연구를 수행할 수 있다(Ruppert et al., 2019; Xing et al., 2022). 과거에는 주로 재래식 모니터링 방법을 통해 특정 환경에 서식하는 생물들을 직접 채집하여 해당 환경의 생물 구성 및 종 다양성 연구를 수행하였다. 하지만 서식하는 모든 생물을 채집하기에는 한계가 있어 다양한 생물 종을 조사할 경우 시간과 비용 측면에서 비효율적이며, 채집 과정의 위험성과 환경 파괴 가능성이 높다(Thomsen and Willerslev, 2015). 반면 eDNA는 높은 민감도를 가져 다양한 생물의 종 동정이 가능하고, 직접 생물을 채집하는 방법에 비해 상대적으로 안전할 뿐 아니라 환경 교란을 최소화할 수 있으며, 조사 시간과 비용이 절감된다는 다양한 장점이 있다(Smart et al., 2015; Deiner et al., 2017).
최근에는 이러한 eDNA의 특성을 활용하여 다양한 환경의 생태계 모니터링 연구가 활발히 수행되고 있으며, 특히 해수로부터 얻은 eDNA를 기반으로 해양생태계를 분석하는 연구가 진행되고 있다(Rees et al., 2014). 국내에서는 동해의 6 지점에서 채집한 해수의 eDNA metabarcoding 분석을 통해 해당 지역에 서식하는 식물플랑크톤의 군집구조를 분석하고 종 다양성을 평가한 연구 결과가 존재한다(Yoon et al., 2016). 또한 국내 남해안 연안의 해수와 퇴적물의 eDNA를 이용한 metagenome 분석을 통해 미생물 군집구조를 파악하여 인위적으로 발생한 환경오염이 미친 영향을 연구한 바가 있다(Won et al., 2017). 해외에서도 eDNA를 활용한 다양한 연구 결과가 존재한다. 미국에서는 뉴저지 연안에 서식하는 해양어류의 계절 별 다양성 분석을 위해 트롤망을 사용하여 어류를 채집하는 방법과 eDNA를 활용하여 분석하는 방법을 비교하였으며, eDNA를 활용한 분석법이 더 효율적이고 정확한 결과를 도출했음을 밝혔다(Stoeckle et al., 2021). 또한 대서양과 태평양에서 채집한 해수를 기반으로 eDNA metabarcoding 분석을 통해 해양에 서식하는 작은 동물플랑크톤의 종 다양성을 규명한 연구 결과가 보고되었다(Garcia-Vazquez et al., 2021). 더 나아가 eDNA는 극지나 심해와 같이 극한 환경 조건에 서식하는 다양한 생물들의 모니터링 연구에도 활발히 활용되고 있다(Cowart et al., 2018; Kawato et al., 2021; Jensen et al., 2022).
이처럼 해양 환경으로부터 얻은 eDNA를 기반으로 한 수생태계 모니터링을 통해 다양한 연구를 수행할 수 있다. 특히 최근에는 차세대 염기서열 분석 기법(NGS: Next Generation Sequencing)을 적용한 eDNA 분석 기술이 빠르게 발전하고 있으며 생물 유전 정보 데이터베이스 또한 계속해서 업데이트가 되고 있어 더욱 빠르고 정확한 분석이 가능하다(Berry et al., 2020; Othman et al., 2023). 다양한 생태계를 효율적으로 분석하고 파악하기 위해서는 eDNA 연구가 필연적이며, 정확한 eDNA 연구를 수행하기 위해서는 전반적인 연구 과정에 대한 이해를 필요로 한다. 본 리뷰는 해양생물 다양성 연구를 위해 해양 환경에서 해수 시료를 채집하고, 이로부터 분자생물학적 기법을 통해 추출한 eDNA를 기반으로 Metagenome 분석을 수행한 결과의 해석까지 연구의 전반적인 과정에 대해 살펴보고자 한다.
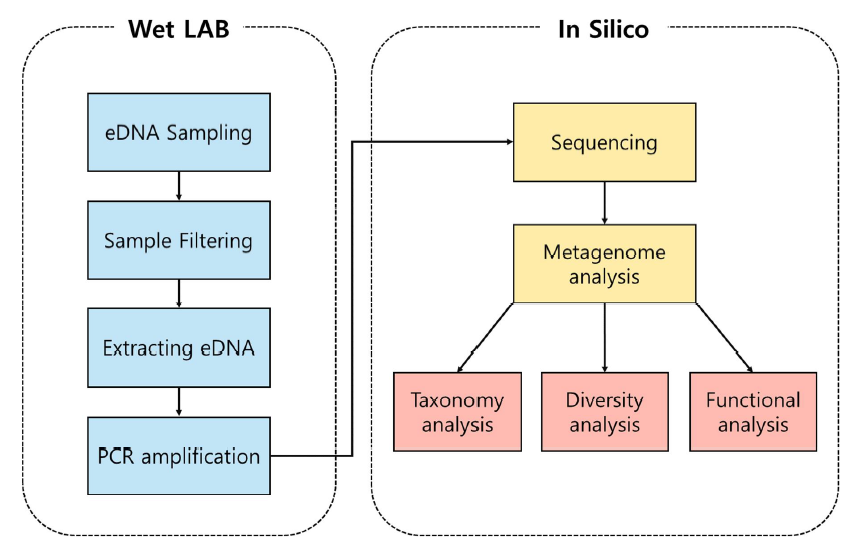
2. eDNA 연구 방법
eDNA를 활용한 해양생물 다양성 연구의 시작은 특정 해양 환경으로부터 해수 시료를 채집하고, 이로부터 eDNA를 추출하는 것이다. 이후 분석을 위해 NGS 기법을 적용할 수 있도록 분자생물학적 기법을 통한 실험 처리 과정을 거쳐야 한다(Fig. 1). 본 리뷰는 eDNA 연구를 위해 환경 시료를 채집하는 방법과 시료로부터 eDNA를 추출하는 방법, 그리고 추출된 DNA를 증폭하고 NGS 기법을 적용하여 eDNA 염기서열을 분석하여 생물 종을 동정하는 방법에 대해 설명하고자 한다.
2.1 해수 시료의 채집
해양환경으로부터 해수를 채집하기 위해서는 먼저 채집 지점 및 정점과 깊이 등을 설정하여야 한다. 해수 채집은 연안이나 해양 한가운데, 또는 심해까지 매우 다양한 환경에서 수행할 수 있다. 연구 목적에 따라 채집 환경도 달라지며, 해수 시료의 채집 방법에도 차이가 생길 수 있다(Fig. 2). 예를 들어, 해수 표면과 같이 얕은 해양 환경에 서식하는 생물 군집을 분석하기 위해서는 채집자가 직접 채수병을 사용하여 표층 해수를 채집할 수 있다. 하지만 심해처럼 수심이 깊은 곳의 해수를 채집하기 위해서는 NiskinTM bottle이나 CTD Rosette와 같은 장비를 사용해야 한다(Suter et al., 2016; Kanhai et al., 2018).

채집하는 해수의 양은 연구 목적과 채집 방법에 따라 수십 mL부터 수십 L까지 다양하지만, 주로 한 지점 당 1~2 L의 해수를 채집한다(Xing et al., 2022). 일반적으로 멸균된 채수병이나 sodium hypochlorite로 내부를 세척한 광구병 등을 사용하여 2 L 가량의 해수를 채집할 수 있다. 채집 과정 동안 외적인 요인에 의해 오염이 일어나지 않도록 주의하며, 특히 채집자의 DNA가 시료에 들어가지 않도록 채집 과정 동안 멸균글러브를 착용하거나, 해수 시료와 채수병 입구에 채집자의 신체 부위 및 외부 물질이 닿지 않도록 유의하여야 한다(Cowart et al., 2022).
채집한 해수는 정확한 eDNA 추출을 위해 채집 직후 바로 여과를 진행하는 것이 좋지만, 현장 여과가 불가능할 경우에는 -20℃에서 -80℃의 냉각 상태로 보관하여 eDNA의 손상 및 분해를 지연시킬 수 있다. 해수 시료는 암조건의 4℃에서 최대 2일까지 보존할 수 있으나, 이후에는 eDNA가 급속도로 분해되어 추후에 분석이 불가능한 상태로 변질될 수 있다(Kim et al., 2021a). 추가적으로, 채집한 해수 시료에 Longmire's 용액(100 mM Tris, 100 mM EDTA, 10 mM NaCl, 0.5% SDS, 0.2% sodium azide)과 같은 특수 보존용액이나 에탄올과 아세트산 나트륨의 혼합용액과 같은 고정용액을 첨가하여 eDNA의 변질을 지연시키는 방법도 존재한다(Williams et al., 2016; Ladell et al., 2018).
2.2 해수 시료의 여과(Filtering)
채집한 해수 시료로부터 eDNA를 추출하기 위해서는 여과 과정을 거쳐야 한다. 여과 시에는 주로 진공 펌프를 사용하며, 여과를 거친 여과지로부터 eDNA를 추출할 수 있다. 일반적으로 많은 양의 해수 시료를 여과할수록 추출되는 eDNA의 수율과 농도가 증가하지만, 연구 목적에 따라 적절한 양의 해수 시료를 여과하는 것이 좋다(Xing et al., 2022). 한 연구에서는 15 mL 부터 2 L 사이의 적은 양의 해수 시료로부터 eDNA를 추출하였을 때, 2 L의 해수 시료로부터 가장 많은 양의 eDNA가 검출되었음을 밝혔다(Muha et al., 2019). 최근 많은 연구에서는 2 L 가량의 해수 시료를 기반으로 해양생물 다양성 분석을 수행하고 있다(Juhel et al., 2020; Sigsgaard et al., 2020; Suter et al., 2021).
공극 크기(pore size)와 재질에 따른 여과지의 종류도 eDNA의 추출에 영향을 미친다. 해수 시료를 여과하기 위해 사용되는 여과지의 공극 크기는 0.22부터 5 μm까지 매우 다양하다. 일반적으로 해양어류의 다양성 분석에는 0.22 또는 0.45 μm 공극 크기의 여과지가 사용되지만, 0.22 μm의 여과지로부터 더 광범위한 범위의 eDNA를 얻을 수 있었다는 연구 결과가 존재한다(Kawakami et al., 2023). 해수 시료 용으로 주로 사용되는 여과지 종류로는 크게 membrane 여과지와 유리섬유 여과지(glass fiber/glass micro- fiber: GF)가 있다. Membrane 여과지는 재질에 따라 CA (cellulose acetate), CN (cellulose nitrate), MCE (mixed cellulose acetate and nitrate) 등으로 나뉠 수 있으며, 유리섬유 여과지는 공극 크기에 따라 GF/F (0.7 μm), GF/C (1.2 μm), GF/D (2.7 μm) 등으로 나뉜다. 이외에도 PES (polyethersulfone), PC (polycarbonate), PVDF (poly- vinylidene fluoride)와 같은 필름 재질의 membrane 여과지도 존재하지만, 이들보다 CN 여과지를 사용한 경우에 eDNA의 수율이 더 높았음을 보인 연구 결과가 있다(Majaneva et al., 2018). Membrane 여과지는 공극 크기가 일정하여 수중의 eDNA를 균일하게 확보할 수 있지만, 수중의 입자성 물질로 인해 여과지가 쉽게 막힐 수 있어 여과량이 상대적으로 적다. 여러 층의 유리섬유로 구성된 유리섬유 여과지의 경우, 공극 크기가 일정하지 않아 균일한 양의 eDNA를 확보하기에는 어려움이 있지만 다른 재질의 여과지에 비해 eDNA 수율이 높은 것으로 나타났다(Sanches and Schreier, 2019). 최근에는 filter capsules (Sterivex enclosure filter, GP) 또는 Bottle Top Vacuum Filter와 같은 일체형 여과세트도 많은 연구에 사용되고 있다(Searle et al., 2016; Wong et al., 2020). 이처럼 다양한 종류의 여과지가 존재하므로 연구의 목적과 해수 시료의 상태에 적절한 여과지를 선택하는 것이 중요하다.
2.3 eDNA 추출
해수 시료를 여과한 여과지로부터 분자생물학적 기법을 적용하여 eDNA를 추출할 수 있다. eDNA 추출을 위해 다양한 DNA 추출 kit가 사용될 수 있으며, 그 중에서도 MP Biomedicals사의 FastDNATM SPIN Kit for Soil, Qiagen 사의 DNeasy Blood and Tissue DNA extraction kit, DNeasy PowerSoil DNA Extraction Kit 그리고 DNeasy PowerWater DNA Extraction Kit 등이 주로 사용된다. Kit 종류마다 DNA 추출 방법과 시약에 차이가 있기 때문에 추출되는 eDNA의 수율과 농도 및 순도에 영향을 줄 수 있다(Eichmiller et al., 2016; Shu et al., 2020).
eDNA 추출 과정은 kit마다 조금씩 차이가 있지만, 공통적으로 모든 과정은 크게 파쇄 및 추출(Cell Lysis and crude extraction), 농축 및 정제(Concentration and Purification), 그리고 용출(Elution) 단계로 나눌 수 있다(Kim et al., 2021a). 파쇄 및 추출 단계에서는 해수 시료를 여과한 여과지를 물리적 및 화학적 방법을 사용하여 세포를 파쇄하고 세포 내부로부터 DNA를 추출한다. 이후 대다수의 kit에서는 컬럼과 에탄올 등이 포함된 다양한 buffer를 이용하여 농축과 정제를 거친 뒤, TE 또는 TAE와 같은 DNA elution buffer를 사용하여 eDNA를 용출하는 방법을 주로 사용하고 있다. 이외에도 kit에서 제시하는 방법을 변형하여 PCI (Phenol-Chloroform-Isoamyl), CTAB (Cetyltrimethylammonium bromide) 등의 용액을 활용한 방법을 통해 eDNA를 추출할 수 있다(Hunter et al., 2019).
2.4 PCR을 통한 유전자 증폭
추출된 eDNA를 기반으로 종 특이적 마커(species-specific marker)를 사용하여 PCR (Polymerase chain reaction)을 수행하면 분석하고자 하는 생물 종의 특정 유전자 부위만을 증폭할 수 있다(MacDonald and Sarre, 2017). 현재까지 많은 eDNA 연구에서 다양한 종 특이적 마커를 사용하여 특정 해양생물 종의 존재 유무를 확인하였고, 더 나아가 멸종위기종이나 외래종 등을 탐지하기 위한 프라이머(primer)도 개발되고 있다(Knudsen et al., 2019; Kim et al., 2021b). 더욱 정확한 종 탐지를 위해 qPCR (real-time quantitative PCR)이나 ddPCR (droplet digital PCR)과 같은 다양한 PCR 방법이 적용되기도 하며, 해양 척추동물을 규명하기 위해 발전된 nested PCR 기법인 'GoFish'와 같이 새로운 PCR 기법들도 제시되고 있다(Stoeckle et al., 2018). 하지만 종 특이적 마커는 하나의 생물 종에만 초점을 맞추어 특정 종의 유전 정보만 특이적으로 증폭할 수 있는 프라이머이므로, 이를 생물 다양성 연구에 적용하기에는 어려움이 있다.
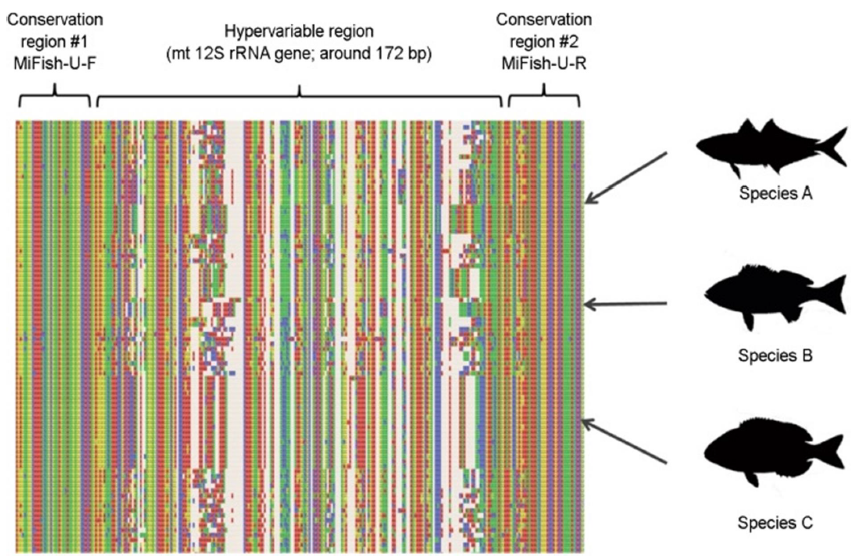
eDNA로부터 다양한 생물을 분석하기 위해서는 여러 생물 종의 유전자를 동시에 증폭할 수 있는 유니버설 프라이머(universal primer)가 필요하다(Stat et al., 2017). 모든 생물에는 종을 구분할 수 있는 종간 변이 염기서열 부위가 존재하는데, 이를 hyper- variable region이라고 한다. Hypervariable region의 앞뒤로는 모든 종에 공통적으로 보존된 염기서열이 존재하며, 이 부위를 conservative region이라고 한다. Universal primer을 사용하면 이러한 conservative region의 서열을 타겟하여 hypervariable region을 증폭시킬 수 있다(Fig. 3). 유니버설 프라이머의 종류에 따라 타겟하는 서열 위치가 다르며, 생물마다 분석을 위해 주로 타겟하는 서열 부위가 다르기 때문에 다양한 종류의 유니버설 프라이머가 생물 다양성 분석에 사용된다. 예를 들어 미생물을 분석하는 경우에는 주로 16S rRNA 지역을 타겟하며, 그 중에서도 V3, V4 region을 증폭하기 위한 유니버설 프라이머가 주로 사용된다(James, 2010; Takahashi et al., 2014). 어류 및 해양 척추동물의 경우에는 미토콘드리아의 12S rRNA 지역을 타겟하는 유니버설 프라이머 세트인 'MiFish-U/E'가 주로 사용되고 있다(Miya et al., 2015). 이외에도 동물플랑크톤 및 해양 무척추동물과 같은 진핵생물의 분석에는 18S rRNA 또는 미토콘드리아의 COI (cytochrome c oxidase I) 지역을 주로 타겟하며(Djurhuus et al., 2018; Giebner et al., 2020), 식물플랑크톤 및 조류를 분석하는 경우에는 23S rRNA 지역을 타겟하는 경우가 많다(Sherwood and Presting, 2007; Yoon et al., 2016). 이처럼 유니버설 프라이머를 활용하여 특정 환경에 존재하는 다양한 생물상을 규명하는 메타바코딩(metabarcoding) 분석 기법은 해양생물 다양성 연구에 활발히 적용되고 있다.
2.5 NGS를 이용한 환경유전체 분석
PCR을 통해 증폭된 특정 서열은 NGS 기법을 기반으로 한 염기서열 분석, 즉 시퀀싱을 수행할 수 있다. eDNA 시퀀싱에는 주로 short-read 서열 분석 장비인 Illumina (https://www.illumina.com) 사의 Miseq, NovaSeq, Hiseq, NextSeq 등과 Thermo Fisher (https:// www.thermofisher.com) 사의 Ion Torrent sequencer가 사용되고 있다(Stat et al., 2017; Peters et al., 2018; Singer et al., 2019; Qu et al., 2020). Long-read 서열 분석 시에는 Pacific Biosciences (https://www.pacb.com) 또는 Oxford Nanopore Technologies (https://nanoporetech.com) 사의 분석 기술을 적용하기도 한다(Patin and Goodwin, 2022). PCR로 증폭된 염기서열 길이에 따라 시퀀싱하는 유전자 서열 길이가 달라지므로 상황에 맞는 서열 분석 장비를 선택해야 한다. 또한 시퀀싱을 수행하기 전에는 분석 장비에 적합한 어댑터(adapter) 및 인덱스(index)를 해당 염기서열에 부착하여 시퀀싱이 가능한 형태의 라이브러리(library)를 제작하여야 한다(Head et al., 2014).
시퀀싱을 통해 얻은 염기서열 조각들을 유전자 정보 데이터베이스(database)를 기반으로 검색하면 해당 서열에 대한 종 동정을 진행할 수 있다. 현재까지도 여러 해양생물의 종 동정을 위해 다양한 레퍼런스 데이터베이스가 활용되고 있으며, 데이터베이스 내의 유전 정보도 계속해서 업데이트되고 있다(Mineta and Gojobori, 2016). 가장 대중적으로 활용되는 유전 정보 데이터베이스인 NCBI (National Center for Biotechnology Information, https://www.ncbi.nlm.nih.gov) 외에도 rRNA 유전 정보들을 기반으로 한 SILVA (from Latin silva, forest, https://www.arb-silva.de), 다양한 유전체 시퀀싱 프로젝트와 관련된 메타데이터를 다루는 GOLD (Genomes Online Database, https://gold.jgi.doe.gov), 대규모의 진핵생물 DNA 바코드 정보를 제공하는 BOLD (Barcode od Life Database, https://boldsystems.org) 등이 eDNA 연구에 활용되고 있다(Lacoursiere-Roussel et al., 2018; Mukherjee et al., 2021; Kwak et al., 2022; Minardi et al., 2022).
3. eDNA 기반 Metagenome 연구
eDNA는 대체로 특정 환경에 서식하는 다양한 생물 종의 유전 정보가 포함되어 있다. 앞서 언급한 메타바코딩 기법을 통해 eDNA로부터 미생물부터 척추동물까지 포괄적인 생물 군집의 다양성을 규명할 수 있다. 반면, 다양한 genome의 집합을 Metagenome이라고 하는데, 이처럼 eDNA를 기반으로 한 Meta- genome 연구를 통해 특정 환경에 서식하는 생물의 분류학적 분석 및 생물 종 다양성 분석이 가능하다. 해양생물 다양성 연구에도 해수 시료를 기반으로 하여 Metagenome 연구를 수행할 수 있다. Metagenome 연구는 생물정보학적 파이프라인(bioinfor- matics pipeline)을 활용하여 시퀀싱 데이터를 소프트웨어 알고리즘을 기반으로 분석하고, 분석 결과를 분류학적 및 다양성 측면과 더 나아가 유전자 기능적 차원에서 해석하는 과정으로 수행된다.
3.1 생물정보학적 파이프라인(Bioinformatics pipe- line)을 활용한 Metagenome 분석
Metagenome 분석을 위해 주로 활용되는 파이프라인으로는 QIIME 2 (Bolyen et al., 2019)를 포함하여, Phylopythia (McHardy et al., 2007), Mothur (Schloss et al., 2009), Usearch (Alloui et al., 2015), OBITools (Boyer et al., 2016) 등이 있다. 특히 QIIME 2는 다양한 운영 체제에서 실행이 가능하기 때문에 Metagenome 분석에 자주 사용된다(Kuczynski et al., 2012). 파이브라인마다 분석 과정에 조금씩 차이가 있지만 일반적인 Metagenome 분석 과정은 raw 시퀀스 데이터로부터 Demultiplexing, Denoising, Clustering 그리고 Taxonomy classification 단계로 나뉠 수 있다(Liu et al., 2021; Xing et al., 2022). Demultiplexing은 각각의 시퀀싱 read가 어떤 샘플로부터 유래했는지 나누는 단계이며, Denoising 단계에서는 오류 및 불필요한 서열들을 제거하는 quality filtering을 수행한다. 이후 Clustering 단계에서는 filtering된 read를 OTUs (Operational Taxonomic Units) 또는 ASVs (Amplicon Sequence Variants) 단위로 묶어준다. 마지막으로 Taxonomy classification 단계에서는 앞서 형성된 OTUs 또는 ASVs를 특정 유전 정보 데이터베이스의 분류군과 비교하여 분류학적인 종 동정을 수행한다. 파이프라인과 데이터베이스의 종류에 따라 Metagenome 분석 결과가 달라질 수 있으며, 분석 결과를 해석하는 방법도 다양하기 때문에 연구 목적에 맞는 분석 방법을 적용하는 것이 중요하다.
3.2 Metagenome 분석 결과의 해석
생물정보학적 파이프라인을 통해 얻은 Metagenome 분석 결과로 다양한 생물 종을 규명할 수 있을 뿐 아니라, 결과 데이터를 활용하여 분류학적 차원에서의 생물 종 동정과 다양성 분석, 더 나아가 유전자의 기능적 차원에서의 분석과 같은 추가적인 분석이 가능하다(Liu et al., 2021).
3.2.1 Taxonomy analysis
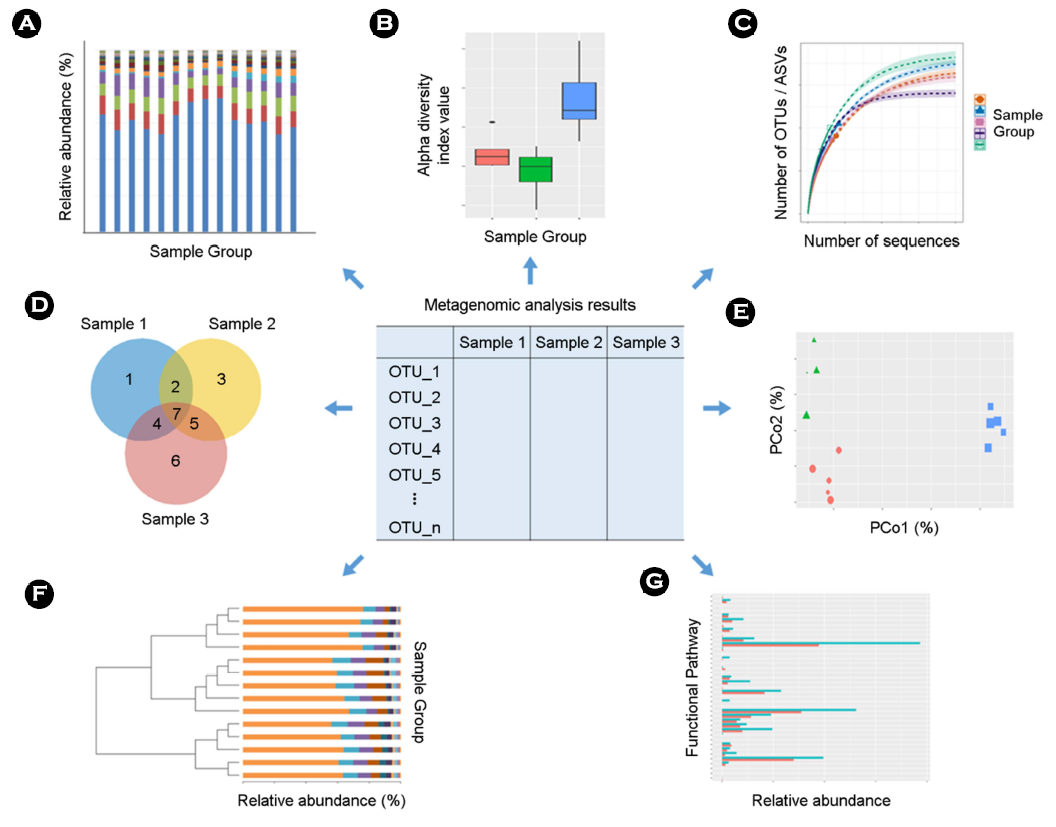
Metagenome 분석 결과 중 데이터베이스를 기반으로 한 종 동정 결과를 활용하여 eDNA에 존재하는 다양한 유전 정보가 각각 어떤 생물 종에 해당하는 것인지를 분류학적으로 확인할 수 있다. 일반적으로 생물분류 기본 단위를 기반으로 구분하여 계(kingdom)부터 종(species)까지의 생물 종 동정이 가능하다. 분류학적 분석 결과는 특정 분류 단위에 따라 시료 별 또는 그룹 별 누적 막대 그래프로 나타낼 수 있으며, 이를 통해 생물 종 구성을 확인할 수 있다(Van Rossum et al., 2016). 일반적으로 상대적인 종 구성 비율을 비교하기 위해 100% 기준 비율 누적 그래프를 통해 결과를 나타내며, 절대적인 구성 종의 수를 확인하려면 비율 그래프가 아닌 개수 누적 그래프로 확인할 수 있다(Fig. 4A).
3.2.2 Diversity analysis
생물 다양성 연구에 적용되는 분석법은 크게 시료 내 생물 종 다양성을 분석하는 Alpha diversity 분석과, 시료 간 생물 다양성의 유사도를 비교 분석하는 Beta diversity 분석으로 나뉠 수 있다(Calle, 2019). Metagenome 분석 결과를 통계학적 처리를 통해 다양성 분석 결과를 나타낼 수 있으며, 결과를 시각적으로 나타내기 위해 R 및 RStudio (https://www.r-project.org/)와 같은 프로그램을 사용하여 plot을 그릴 수 있다.
(1) Alpha diversity analysis
Alpha diversity 분석에서는 다양한 Alpha diversity index를 활용하여 시료 내의 종 풍부도(sample richness) 및 생물 종 분포의 균등도(evenness)를 평가한다. 주로 사용되는 Alpha diversity index로는 종 풍부도를 평가할 수 있는 Observed Features (species or OTUs), Chao1, Faith's PD (Phylogenetic Diversity), ACE (Abundance-based Coverage Estimators) 등과 균등도를 함께 평가할 수 있는 Simpson index, Shannon index, 그리고 Pielou's evenness 등이 적용되고 있다(Liu et al., 2018; Zhao et al., 2022). Metagenome 분석 결과로 시료 별 각 alpha diversity index에 대한 값(value)를 기반으로 특정 그룹 별로 묶어 alpha diversity 값에 대한 box plot 형태로 나타낼 수 있으며, 그룹 별 비교가 불가능할 경우 alpha diversity value의 값 자체를 시료 별로 비교하기도 한다(Fig. 4B).
또한 Good's coverage index 및 Rarefaction curve를 통해 시퀀스 수에 따른 생물 다양성 변화를 평가할 수 있다(Zhou et al., 2020). Good's coverage 값이 1.0, 즉 100%에 가까울수록, 또는 Rarefaction curve가 어느 한계점 이상에서 완만 곡선을 그리는 형태를 보일수록 시퀀스의 양이 생물 종의 군집 분포를 나타낼 수 있을 정도로 충분하다는 분석 결과의 신뢰성을 확인할 수 있다(Fig. 4C).
다양한 alpha diversity를 기반으로 다양성 분석을 하는 것 외에도, Metagenome 분석 파이프라인을 통해 분류된 OTU 또는 ASV 개수를 기반으로 벤 다이어그램을 통해 시료 별 OTU 및 ASV 풍부도(abundance)를 나타낼 수 있다(Fig. 4D). 이를 통해 시료마다 고유하게 갖는 OTU 및 ASV의 개수와 시료 별로 중복되어 관찰되는 개수는 몇 개인지 직관적으로 파악이 가능하다.
(2) Beta diversity analysis
Beta diversity 분석에서는 시료 간의 유사도 거리를 측정하여 그 다양성을 비교 분석 할 수 있다. Classical scaling이라고도 알려진 PCoA (Principal Coordinates Analysis)를 통해 Beta diversity 분석 결과를 scatter plot으로 나타낼 수 있다(Fig. 4E). 이때 시료 간 유사도 거리 측정을 위해 다양한 distance index가 사용되며, 일반적으로 Metagenome 분석 결과를 나타내기 위해 활용되는 index로는 Jaccard, Bray-Curtis, Unweighted UniFrac 그리고 Weighted UniFrac 등이 있다(Liddicoat et al., 2022). Jaccard와 Unweighted UniFrac의 경우 시료 간 공유하는 생물 종의 유무를 확인하며, Bray-Curtis와 Weighted UniFrac은 생물 종의 유무와 풍부도까지 고려하여 유사도 거리를 측정한다. 또한 Unweighted UniFrac과 Weighted UniFrac은 Phylogenetic tree에 기반하여 유사도 거리를 측정한다는 점에서 Jaccard, Bray-Curtis와 차이가 있다.
Beta diversity 분석에 사용되는 유사도 거리 측정 index를 기반으로 UPGMA tree (Unweighted Pair Group Method with Arithmetic Mean)를 그릴 수 있다(Wang et al., 2018). UPGMA Tree는 분석하고자 하는 분류군 중 가장 가까운 관계끼리 하나의 군으로 묶어 나가는 직관적인 유사도 거리 계산을 반복하여, 최종적으로 얻은 계통 관계도는 서로 유사한 특징을 가질수록 더 가까이 연관되어 있다. UPGMA Tree와 시료 별 분류학적 생물 종 구성 그래프를 함께 묶어 분석 결과를 나타낸다면 직관적으로 시료 별 유사도와 함께 생물 구성 비교도 가능하다(Fig. 4F).
3.2.3 Functional analysis
대부분의 eDNA를 활용한 Metagenome 분석은 분류학적 분석과 생물 다양성 분석에 초점이 맞춰져 있지만, 생태계와 생물 간의 상호작용을 파악하기 위해 기능적 분석도 수행할 수 있다. 일반적으로 eDNA 내 유전 정보를 기반으로 KEGG (Kyoto Encyclo- pedia of Genes and Genomes)나 COG (Clusters of Orthologous Groups of proteins) 데이터베이스를 활용하여 대사 활동 네트워크 분석을 수행할 수 있다(Raiyani and Singh, 2020). 특히 KEGG 경로는 유전자 산물 간의 상호작용을 예측하여 수많은 분자 간의 상호작용과 대사 활동을 나타내는 네트워크 도표로, Metagenome 기능 분석에 흔히 사용된다. KEGG 데이터베이스 분석 결과는 PICRUSt (Phylogenetic Investigation of Communities by Recon- struction of Unobserved States)와 같은 소프트웨어를 사용하여 특정 레벨을 기준으로 관찰된 KEGG 경로의 상대적인 비율을 시료 별로 막대 그래프 형태로 나타낼 수 있다(Fig. 4G).
4. 결론
특정 해수 시료로부터 얻은 eDNA를 활용하면 해당 지역에 서식하는 해양생물들의 종 다양성을 파악할 수 있다. eDNA를 활용한 해양생물 다양성 연구는 현재까지도 활발히 진행되고 있다. 더 효율적인 연구를 위해 eDNA 분석에 활용되는 각종 실험 및 분석 기기도 개발되고 있으며, 분석에 활용되는 각종 파이프라인과 데이터베이스 또한 계속해서 업데이트 되고 있어 더욱 빠르고 정확한 분석이 가능해지고 있다.
하지만 eDNA 분석 연구에도 한계점이 존재한다(Burian et al., 2021). 직접적인 생물 종의 관찰에 의존하지 않고 간접적으로 그 존재를 확인할 수 있기 때문에, 이미 해당 환경에 서식하지 않는 특정 생물이 과거에 분비한 DNA가 검출될 수 있다. 또한 민감도가 높기 때문에 채수 및 실험 과정에서 오염에 의한 오류가 발생할 확률이 높다. 생물적인 요인 외에도 수온, 일조량 그리고 유속과 같은 물리적인 변수도 eDNA에 영향을 줄 수 있다. 따라서 이러한 한계점을 인지하고 이를 보완하여 정확한 분석을 위해 다양한 변수의 보정이 필요하다.
아직까지 국내에서는 보편화된 eDNA 활용 해양생물 다양성 연구 방법이 존재하지 않는다. 연구의 목적에 따라 차이점이 있을 수 있지만 연구 과정에서 다양한 변수에 따른 오류를 최소화하기 위해서는 시료 채집부터 실험과 분석 과정의 통일성이 필요하다. 본 리뷰는 국내 해양 지역에 보편적으로 적용할 수 있는 eDNA를 활용한 해양생물 다양성 연구 방법의 정립에 대한 기초 자료로 활용될 수 있다. 아직까지 생태계 모니터링이 이루어지지 않은 국내 해양 환경에 이러한 eDNA 연구 방법을 적용하여 해양생물 다양성을 파악할 수 있을 것이다.
- References
-
1. Alloui T, Boussebough I, Chaoui A, Nouar AZ, Chettah MC. 2015. Usearch: A meta search engine based on a new result merging strategy. In: International Joint Conference on Knowledge Discovery, Knowledge Engineering and Know- ledge Management (IC3K), Lisbon, Portugal, 12-14 Nov, 2015, pp 531-536.
-
2. Barnes MA, Turner CR, Jerde CL, Renshaw MA, Chadderton WL, Lodge DM. 2014. Environmental conditions influence eDNA persistence in aquatic systems. Environmental Science & Technology 48: 1819-1827.
-
3. Berry O, Jarman S, Bissett A, Hope M, Paeper C, Bessey C, Schwartz MK, Hale J, Bunce M. 2020. Making environmental DNA (eDNA) biodiversity records globally accessible. Environmental DNA 3: 699-705.
-
4. Bolyen E, Rideout JR, Dillon MR, Bokulich NA, Abnet CC, Al-Ghalith GA, Alexander H, Alm EJ, Arumugam M, Asnicar F, et al. 2019. Reproducible, interactive, scalable and exten- sible microbiome data science using QIIME 2. Nature Bio- technology 37: 852-857.
-
5. Boyer F, Mercier C, Bonin A, Le Bras Y, Taberlet P, Coissac E. 2016. obitools: a unix-inspired software package for DNA meta- barcoding. Molecular Ecology Resources 16: 176-182.
-
6. Burian A, Mauvisseau Q, Bulling M, Domisch S, Qian S, Sweet M. 2021. Improving the reliability of eDNA data interpretation. Molecular Ecology Resources 21: 1422-1433.
-
-
8. Cowart DA, Murphy KR, Cheng CHC. 2018. Metagenomic sequen- cing of environmental DNA reveals marine faunal assem- blages from the West Antarctic Peninsula. Marine Genomics 37: 148-160.
-
9. Cowart DA, Murphy KR, Cheng CHC. 2022. Environmental DNA from Marine Waters and Substrates: Protocols for Sampling and eDNA Extraction. Methods in Molecular Biology 2498: 225-251.
-
10. Deiner K, Bik HM, Machler E, Seymour M, Lacoursiere-Roussel A, Altermatt F, Creer S, Bista I, Lodge DM, de Vere N, et al. 2017. Environmental DNA metabarcoding: Transforming how we survey animal and plant communities. Molecular Ecology 26: 5872-5895.
-
11. Djurhuus A, Pitz K, Sawaya NA, Rojas-Marquez J, Michaud B, Montes E, Muller-Karger F, Breitbart M. 2018. Evaluation of marine zooplankton community structure through environ- mental DNA metabarcoding. Limnology Oceanography: Methods 16: 209-221.
-
12. Eichmiller JJ, Miller LM, Sorensen PW. 2016. Optimizing techniques to capture and extract environmental DNA for detection and quantification of fish. Molecular Ecology Resources 16: 56-68.
-
13. Garcia-Vazquez E, Georges O, Fernandez S, Ardura A. 2021. eDNA metabarcoding of small plankton samples to detect fish larvae and their preys from Atlantic and Pacific waters. Scientific Reports 11: 7224.
-
14. Giebner H, Langen K, Bourlat SJ, Kukowka S, Mayer C, Astrin JJ, Misof B, Fonseca VG. 2020. Comparing diversity levels in environmental samples: DNA sequence capture and meta- barcoding approaches using 18S and COI genes. Molecular Ecology Resources 20: 1333-1345.
-
15. Head SR, Komori HK, LaMere SA, Whisenant T, Van Nieuwerburgh F, Salomon DR, Ordoukhanian P. 2014. Library construction for next-generation sequencing: overviews and challenges. Biotechniques 56: 61-77.
-
16. Hunter ME, Ferrante JA, Meigs-Friend G, Ulmer A. 2019. Improving eDNA yield and inhibitor reduction through increased water volumes and multi-filter isolation techniques. Scientific Reports 9: 5259.
-
17. James G. 2010. Universal Bacterial Identification by PCR and DNA Sequencing of 16S rRNA Gene. Shuller M (eds.), PCR for Clinical Microbiology, Springer, Dordrecht, pp 209-214.
-
18. Jensen MR, Høgslund S, Knudsen SW, Nielsen J, Møller PR, Rysgaard S, Thomsen PF. 2022. Distinct latitudinal com- munity patterns of Arctic marine vertebrates along the East Greenlandic coast detected by environmental DNA. Diversity and Distributions 29: 316-334.
-
19. Juhel JB, Utama RS, Marques V, Vimono IB, Sugeha HY, Kadarusman, Pouyaud L, Dejean T, Mouillot D, Hocde R. 2020. Accumulation curves of environmental DNA sequences predict coastal fish diversity in the coral triangle. Proceedings of the Royal Society B 287: 20200248.
-
20. Kanhai LDK, Gardfeldt K, Lyashevska O, Hassellov M, Thompson RC, O'Connor I. 2018. Microplastics in sub-surface waters of the Arctic Central Basin. Marine Pollution Bulletin 130: 8-18.
-
21. Kawakami T, Yamazaki A, Asami M, Goto Y, Yamanaka H, Hyodo S, Ueno H, Kasai A. 2023. Evaluating the sampling effort for the metabarcoding-based detection of fish environmental DNA in the open ocean. Ecology and Evolution 13: e9921.
-
22. Kawato M, Yoshida T, Miya M, Tsuchida S, Nagano Y, Nomura M, Yabuki A, Fujiwara Y, Fujikura K. 2021. Optimization of environmental DNA extraction and amplification methods for metabarcoding of deep-sea fish. MethodsX 8: 101238.
-
23. Kim K, Ryu J, Hwang S. 2021a. Sampling and Extraction Method for Environmental DNA (eDNA) in Freshwater Ecosystems. Korean Journal of Ecology and Environment 54: 170-189.
-
24. Kim P, Yoon T-J, Shin S. 2021b. Monthly eDNA Monitoring of an Invasive Bryozoan, Bugulina californica, in Seawater Using Species-Specific Markers. Animals 11: 1-18.
-
25. Knudsen SW, Ebert RB, Hesselsøe M, Kuntke F, Hassingboe J, Mortensen PB, Thomsen PF, Sigsgaard EE, Hansen BK, Nielsen EE, et al. 2019. Species-specific detection and quantification of environmental DNA from marine fishes in the Baltic Sea. Journal of Experimental Marine Biology and Ecology 510: 31-45.
-
26. Kuczynski J, Stombaugh J, Walters WA, Gonzalez A, Caporaso JG, Knight R. 2012. Using QIIME to analyze 16S rRNA gene sequences from microbial communities. Current Protocols in Microbiology 27: 1E-5.
-
27. Kwak I-S, Ji CW, Kim W-S, Kong D. 2022. The List of Korean Organisms Registered in the NCBI Nucleotide Database for Environmental DNA Research. Korean Journal of Ecology and Environment 55: 352-359.
-
28. Lacoursiere-Roussel A, Howland K, Normandeau E, Grey EK, Archambault P, Deiner K, Lodge DM, Hernandez C, Leduc N, Bernatchez L. 2018. eDNA metabarcoding as a new surveil- lance approach for coastal Arctic biodiversity. Ecology and Evolution 8: 7763-7777.
-
29. Ladell BA, Walleser LR, McCalla SG, Erickson RA, Amberg JJ. 2018. Ethanol and sodium acetate as a preservation method to delay degradation of environmental DNA. Conservation Genetics Resources 11: 83-88.
-
30. Liddicoat C, Krauss SL, Bissett A, Borrett RJ, Ducki LC, Peddle SD, Bullock P, Dobrowolski MP, Grigg A, Tibbett M, et al. 2022. Next generation restoration metrics: Using soil eDNA bac- terial community data to measure trajectories towards re- habilitation targets. Journal of Environmental Management 310: 114748.
-
31. Liu J, Chen X, Shu HY, Lin XR, Zhou QX, Bramryd T, Shu WS, Huang LN. 2018. Microbial community structure and function in sediments from e-waste contaminated rivers at Guiyu area of China. Environmental Pollution 235: 171-179.
-
32. Liu Y-X, Qin Y, Chen T, Lu M, Qian X, Guo X, Bai Y. 2021. A practical guide to amplicon and metagenomic analysis of microbiome data. Protein Cell 12: 315-330.
-
33. MacDonald AJ, Sarre SD. 2017. A framework for developing and validating taxon-specific primers for specimen identification from environmental DNA. Molecular Ecology Resources 17: 708-720.
-
34. Majaneva M, Diserud OH, Eagle SHC, Bostrom E, Hajibabaei M, Ekrem T. 2018. Environmental DNA filtration techniques affect recovered biodiversity. Scientific Reports 8: 4682.
-
35. McHardy AC, Martin HG, Tsirigos A, Hugenholtz P, Rigoutsos I. 2007. Accurate phylogenetic classification of variable-length DNA fragments. Nature Methods 4: 63-72.
-
36. Minardi D, Ryder D, Del Campo J, Garcia Fonseca V, Kerr R, Mortensen S, Pallavicini A, Bass D. 2022. Improved high throughput protocol for targeting eukaryotic symbionts in metazoan and eDNA samples. Molecular Ecology Resources 22: 664-678.
-
-
38. Miya M, Sato Y, Fukunaga T, Sado T, Poulsen JY, Sato K, Minamoto T, Yamamoto S, Yamanaka H, Araki H, et al. 2015. MiFish, a set of universal PCR primers for metabarcoding environ- mental DNA from fishes: detection of more than 230 subtropical marine species. Royal Society Open Science 2: 150088.
-
39. Muha TP, Robinson CV, Garcia de Leaniz C, Consuegra S. 2019. An optimised eDNA protocol for detecting fish in lentic and lotic freshwaters using a small water volume. PLoS One 14: e0219218.
-
40. Mukherjee S, Stamatis D, Bertsch J, Ovchinnikova G, Sundaramurthi JC, Lee J, Kandimalla M, Chen IA, Kyrpides NC, Reddy TBK. 2021. Genomes OnLine Database (GOLD) v.8: overview and updates. Nucleic Acids Research 49: D723-D733.
-
41. Othman N, Muniar K, Haris H, Ramli FF, Sariyat NH, Najmuddin MF, Abdul-Latiff MAB. 2023. A Review of Next-Generation Wildlife Monitoring using Environmental DNA (eDNA) Detec- tion and Next-Generation Sequencing in Malaysia. Sains Malaysiana 52: 17-33.
-
42. Patin NV, Goodwin KD. 2022. Long-Read Sequencing Improves Recovery of Picoeukaryotic Genomes and Zooplankton Marker Genes from Marine Metagenomes. mSystems 7: e00595-00522.
-
43. Peters L, Spatharis S, Dario MA, Dwyer T, Roca IJT, Kintner A, Kanstad-Hanssen Ø, Llewellyn MS, Praebel K. 2018. Environ- mental DNA: A new low-cost monitoring tool for pathogens in salmonid aquaculture. Frontiers in Microbiology 9: 3009.
-
44. Qu C, Stewart KA, Clemente-Carvalho R, Zheng J, Wang Y, Gong C, Ma L, Zhao J, Lougheed SC. 2020. Comparing fish prey diversity for a critically endangered aquatic mammal in a reserve and the wild using eDNA metabarcoding. Scientific Reports 10: 16715.
-
45. Raiyani NM, Singh SP. 2020. Taxonomic and functional profiling of the microbial communities of Arabian Sea: A metagenomics approach. Genomics 112: 4361-4369.
-
46. Rees HC, Maddison BC, Middleditch DJ, Patmore JRM, Gough KC. 2014. The detection of aquatic animal species using environ- mental DNA - a review of eDNA as a survey tool in ecology. Journal of Applied Ecology 51: 1450-1459.
-
47. Ruppert KM, Kline RJ, Rahman MS. 2019. Past, present, and future perspectives of environmental DNA (eDNA) metabarcoding: A systematic review in methods, monitoring, and applications of global eDNA. Global Ecology and Conservation 17: 1-29.
-
48. Sanches TM, Schreier AM. 2019. Optimizing an eDNA protocol for monitoring endangered Chinook Salmon in the San Francisco Estuary: balancing sensitivity, cost and time. bioRxiv 871368: 1-32.
-
49. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, et al. 2009. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environ- mental Microbiology 75: 7537-7541.
-
50. Searle D, Sible E, Cooper A, Putonti C. 2016. 18S rDNA dataset profiling microeukaryotic populations within Chicago area nearshore waters. Data in Brief 6: 526-529.
-
51. Sherwood AR, Presting GG. 2007. Univeral primers amplify a 23S rDNA plastid marker in eukaryotic algae and cyanobacteria. Journal of Phycology 43: 605-608.
-
52. Shu L, Ludwig A, Peng Z. 2020. Standards for Methods Utilizing Environmental DNA for Detection of Fish Species. Genes 11: 1-14.
-
53. Sigsgaard EE, Torquato F, Froslev TG, Moore ABM, Sorensen JM, Range P, Ben-Hamadou R, Bach SS, Moller PR, Thomsen PF. 2020. Using vertebrate environmental DNA from seawater in biomonitoring of marine habitats. Conservation Biology 34: 697-710.
-
54. Singer GAC, Fahner NA, Barnes JG, McCarthy A, Hajibabaei M. 2019. Comprehensive biodiversity analysis via ultra-deep patterned flow cell technology: a case study of eDNA meta- barcoding seawater. Scientific Reports 9: 5991.
-
55. Smart AS, Tingley R, Weeks AR, van Rooyen AR, McCarthy MA. 2015. Environmental DNA sampling is more sensitive than a traditional survey technique for detecting an aquatic invader. Ecological Applications 25: 1944-1952.
-
56. Stat M, Huggett MJ, Bernasconi R, DiBattista JD, Berry TE, Newman SJ, Harvey ES, Bunce M. 2017. Ecosystem biomonitoring with eDNA: metabarcoding across the tree of life in a tropical marine environment. Scientific Reports 7: 12240.
-
57. Stoeckle MY, Adolf J, Charlop-Powers Z, Dunton KJ, Hinks G, VanMorter SM. 2021. Trawl and eDNA assessment of marine fish diversity, seasonality, and relative abundance in coastal New Jersey, USA. ICES Journal of Marine Science 78: 293-304.
-
58. Stoeckle MY, Das Mishu M, Charlop-Powers Z. 2018. GoFish: A versatile nested PCR strategy for environmental DNA assays for marine vertebrates. PLoS One 13: e0198717.
-
59. Suter EA, Scranton MI, Chow S, Stinton D, Medina Faull L, Taylor GT. 2016. Niskin bottle sample collection aliases microbial community composition and biogeochemical interpretation. Limnology and Oceanography 62: 606-617.
-
60. Suter L, Polanowski AM, Clarke LJ, Kitchener JA, Deagle BE. 2021. Capturing open ocean biodiversity: Comparing environmental DNA metabarcoding to the continuous plankton recorder. Molecular Ecology 30: 3140-3157.
-
61. Taberlet P, Coissac E, Hajibabaei M, Rieseberg LH. 2012. Environ- mental DNA. Molecular Ecology 21: 1789-1793.
-
62. Takahashi S, Tomita J, Nishioka K, Hisada T, Nishijima M. 2014. Development of a prokaryotic universal primer for simultan- eous analysis of Bacteria and Archaea using next-generation sequencing. PLoS One 9: e105592.
-
63. Thomsen PF, Willerslev E. 2015. Environmental DNA – An emerging tool in conservation for monitoring past and present bio- diversity. Biological Conservation 183: 4-18.
-
64. Van Rossum T, Pylatuk MM, Osachoff HL, Griffiths EJ, Lo R, Quach M, Palmer R, Lower N, Brinkman FSL, Kennedy CJ. 2016. Microbiome Analysis Across a Natural Copper Gradient at a Proposed Northern Canadian Mine Site. Frontiers in Environ- mental Science 3: 1-12.
-
65. Wang X, Wang Z, Jiang P, He Y, Mu Y, Lv X, Zhuang L. 2018. Bacterial diversity and community structure in the rhizo- sphere of four Ferula species. Scientific Reports 8: 5345.
-
66. Wilcox TM, McKelvey KS, Young MK, Jane SF, Lowe WH, Whiteley AR, Schwartz MK. 2013. Robust detection of rare species using environmental DNA: the importance of primer specificity. PLoS One 8: e59520.
-
67. Williams KE, Huyvaert KP, Piaggio AJ. 2016. No filters, no fridges: a method for preservation of water samples for eDNA analysis. BMC Research Notes 9: 298.
-
68. Won NI, Kim KH, Kang JH, Park SR, Lee HJ. 2017. Exploring the Impacts of Anthropogenic Disturbance on Seawater and Sediment Microbial Communities in Korean Coastal Waters Using Metagenomics Analysis. International Journal of Environmental Researcn and Public Health 14: 1-17.
-
69. Wong MK-S, Nakao M, Hyodo S. 2020. Field application of an improved protocol for environmental DNA extraction, purifi- cation, and measurement using Sterivex filter. Scientific Reports 10: 21531.
-
70. Xing Y, Gao W, Shen Z, Zhang Y, Bai J, Cai X, Ouyang J, Zhao Y. 2022. A Review of Environmental DNA Field and Laboratory Protocols Applied in Fish Ecology and Environmental Health. Frontiers in Environmental Science 10: 1-17.
-
71. Yoon T-H, Kang H-E, Kang C-K, Lee SH, Ahn D-H, Park H, Kim H-W. 2016. Development of a cost-effective metabarcoding strategy for analysis of the marine phytoplankton community. PeerJ 4: e2115.
-
72. Zhao W, Zhang Z, Gao Y, Liu X, Du C, Ma F, Wang S, Shi W, Yang Y, Deng R, et al. 2022. Fungal dynamic changes in naturally fermented 'Kyoho' grape juice. Archives of Microbiology 204: 556.
-
73. Zhou Z, Yu M, Ding G, Gao G, He Y. 2020. Diversity and structural differences of bacterial microbial communities in rhizo- compartments of desert leguminous plants. PLoS One 15: e0241057.
Copyright (C) 2025. The Korean Society of Marine Life Science