






JMLS 2021 December;6(2):47-57. 10.23005/ksmls.2021.6.2.47 Epub 2021 December 15
Copyright © 2021 by The Korean Society of Marine Life Science
The Application of Genome Research to Development of Aquaculture
Seung Jae Lee; Department of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Jinmu Kim; Department of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Eunkyung Choi; Department of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Euna Jo; Department of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Minjoo Cho; Department of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Hyun Park; Department of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
- Abstract
- 수산업은 수산물의 남획, 국가 간의 제한, 기후변화로 인하여 수산물 개체 수 감소 등으로 전 세계 수산물 생산량이 정체되면서, 양식업은 항상 세계 식량 소비를 위해 오랫동안 지속되어온 패러다임을 뒤집고 인류가 바다, 호수, 강의 풍부함을 새로운 방식으로 확장할 수 있도록 하는 '푸른 혁명'의 가능성을 제시하고 있다. 양식산업은 이제 인간 소비를 위한 해산물의 원료로서 해양에서 얻는 어업생산량의 절반이상을 넘어섰다. 지속 가능한 양식산업의 발전을 위하여 다양한 최신 생물학적 연구 방법들이 적용되고 있다. 유전체학은 2011년 대서양 대구의 염기서열이 규명된 이래 최근 상당한 진전을 이루었으며 더 많은 종의 유전체가 해독되어 우수 형질의 육종 및 번식 기술, 질병 저항성 품질 개량, 양식 사료 및 사료방식의 최적화 등 양식산업을 위한 보다 견고하고 생산적인 지식 기반을 제공한다. 본 리뷰는 수산양식종의 유전체 연구를 위한 유전체 분석 기술과 수산양식종의 유전체 연구의 현황에 대해서 살펴보았다. 이와 같이 유전체 분석 기술과 유전체학의 발전은 수산양식산업의 과제를 해결하는 데 중요하며 지속 가능한 어업과 양식에 도움을 줄 것이다.
In the fishery industry, global aquaculture production has stagnated due to overfishing of aquatic products, restrictions between countries, and climate change. The aquaculture suggests the possibility of a blue revolution that can be expanded in a new way. The aquaculture industry now accounts for more than half of the fishery products from the sea as a raw material for seafood for human consumption. Various latest biological research methods are being applied for the development of a sustainable aquaculture industry. Genomics has made significant progress in recent years. Since the genome sequence of Atlantic cod was sequenced in 2011, the genomes of more species have been sequenced. The genome information is providing a more robust and productive knowledge base for the aquaculture industry, including breeding and breeding of superior traits, improving disease resistance quality, and optimizing aquaculture feed and feed methods. This review looked at the status of genome analysis technology and the current status of genome research of aquaculture species. The development of genome research technology and massive genomic information is important in solving the challenges of the aquaculture industry and will help sustainable fisheries and aquaculture.
Keywords: Aquaculture(양식) Genome(유전체) Transcriptome(전사체) NGS(차세대염기서열 분석)
Correspondence to: Hyun Park;Department of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
- Received
- 13 October 2021;
- Revised
- 15 October 2021;
- Accepted
- 20 October 2021.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Language: Korean/English,
Full Text:

1. 서론
인류는 오랫동안 바다에서 무한한 수산물을 얻는 혜택을 받아왔다. 그러나, 무분별한 남획, 일부 정부의 제한, 기후변화로 추정되는 수산물 개체 수 감소 등으로 전 세계 수산물 생산량이 정체되면서 양식업이 급물살을 타고 있다. 양식업이 기술적으로 발전하고, 규모화되고, 경제성이 높아지면서 양식에 의한 수산물 공급은 1인당 어류 소비 증가를 증대 시켰다. 양식업은 이제 모든 해산물의 약 절반을 생산한다. 실제로 불과 한 세대 전만 해도 상대적으로 미미한 공급에 불과했지만, 2015년 양식업은 인간 소비를 위한 해산물의 원료로서 해양에서 얻는 어업생산량의 절반이상을 넘어섰다. 현재 양식업에서 연간 생산하는 생물량은 쇠고기를 능가하고 있으며, 그 증가율이 가금류 생산량을 넘어 가장 빠르게 성장하고 있는 산업 분야이다. 유엔식량농업기구(FAO)에 따르면 2018년 양식업의 생산량은 1억140만톤에 달하고 매출은 약 2,630억 달러에 달하며, 해양, 담수, 기수 환경에서 수십 종이 사육되고 있고, 향후 2025년에는 2,750억 달러에 이를 것으로 예상된다(FAO, 2018). 특히 국내에서는 김과 미역, 전복, 장어는 국내산 대부분이 양식이고, 송어는 98%, 굴은 89%, 광어는 88%, 새우는 16%가 양식으로 생산되는 등 우리나라는 이미 노르웨이, 덴마크, 일본 등과 더불어 세계 7대 양식업 강국이다.
양식과 어업을 위한 유전체학은 2011년 양식산업의 핵심 어종인 대서양 대구의 염기서열이 최초로 규명(Star et al., 2011)된 이래 지난 10년 동안 상당한 진전을 이루었으며 더 많은 어종의 유전체가 해독되어 이용할 수 있게 되었다. 유전체 정보는 양식 사료 및 사료 급식의 최적화, 번식 기술 또는 유전적 선택 또는 검사(예: 후성유전학, 단백질학 및 대사체학)에 사용될 수 있는 생리학적 연구를 향상시키기 위한 강력한 도구를 제공한다. 유전체 서열은 현재 많은 양식 종에서 사용할 수 있어 insertions/deletions, single nucleotide polymorphisms (SNPs), copy number variations 및 methylated region과 같은 유전체 변이를 식별할 수 있어 생산량의 증대나 고형질의 표현형을 예측하는 데 유용하다. 또한 유전체 기반의 genetic mapping, quantitative trait loci (QTL) analysis, genome-wide association studies (GWAS), expression profiling 등의 연구를 통하여 특정 형질과 관련된 유전형 변형을 식별하는 데 사용될 수 있어 이를 통한 번식 프로그램에 활용될 수 있다. 특히 성장, 번식 및 질병 저항의 기반이 되는 유전자 네트워크에 대한 보다 완벽한 이해는 양식산업을 위한 보다 견고하고 생산적인 유전자를 개발하기 위한 지식 기반을 제공할 것이다. 본 리뷰는 수산양식종의 유전체 연구를 위한 유전체 분석 기술과 수산양식종의 유전체 연구의 현황에 대해서 살펴보고자 한다. 이와 같이 유전체 서열 분석 기술과 유전체학의 발전은 수산양식산업의 과제를 해결하는 데 중요하며 지속 가능한 어업과 양식에 도움이 될 수 있다.
2. 유전체 연구 방법
차세대 염기서열 분석(NGS: Next Generation Sequence) 기술의 발전으로 새로운 종의 유전체를 해독하는 비용과 시간이 획기적으로 줄어들었다. 또한 염기서열 분석에 사용되는 다양한 생물정보학 도구(bioinformatics tools)의 발전으로 유전체 조립(genome assembly), 유전자 예측(gene prediction)과 기능 연구(gene functional analysis)와 같은 분석을 소규모실험실에서도 수행할 수 있게 되었다.
유전체 분석 과정은 염기서열 분석(sequencing), 조립(assembly), 주석(annotation)의 단계로 진행된다(Fig. 1). 염기서열 분석 단계에서는 DNA/RNA 서열 분석을 위하여 NGS 플랫폼을 사용하고, 조립 단계에서 분석된 유전자 서열은 scaffolds를 얻기 위해 조립되며, 이후 주석 단계는 구조 주석(structural annotation)과 기능 주석(functional annotation)을 수행하게 된다. 구조 주석 과정에서는 우선 scaffolds에서 반복되는 서열(repeated sequences)을 masking 하고, 유전자를 찾기 위해 유전자 예측 도구를 사용하면 이들 유전자를 구성하는 인트론(Intron), 엑손(Exon), 비번역 영역(UTR)의 구조가 결정된다. 마지막으로 기능 주석 과정에서는 유전자의 기능을 결정하기 위해 상동 검색(homology search) 및 유전자 온톨로지 맵핑(gene ontology mapping)을 수행한다.
2.1. 서열 분석(Genome Sequencing)
유전자 서열 분석의 정확도는 유전체 조립 결과의 정확도, 유전자 구조 및 기능 분석, 단백질 분석 등에 직접적인 영향을 미친다. NGS 플랫폼에서 생성되는 시퀀스 분석은 읽기 길이(read length), 유형(type), 오류 발생률(error rate) 등 각각의 서열 분석 장비 플랫폼의 다른 특성을 가지고 있다. 유전자 서열 분석 기술로서는 분석되는 유전자 서열의 길이에 따라 구분되어질 수 있다. Short-read 서열 분석 장비로는 Illumina (http://www.illumina.com) 사의 NovaSeq, HiSeq, NextSeq, MiSeq 등과 BGI (https://www.bgi.com)의 MGISEQ과 BGISEQ이 널리 사용되고 있으며 Thermo Fisher (https://www.thermofisher.com)의 Ion Torrent sequencer가 있다. long-read 서열 분석 장비로는 Pacific Biosciences (https://www. pacb.com)의 RSII, Sequel, Sequel II와 Oxford Nanopore (https:// nanoporetech.com)의 MinION, GridION, PromethION가 있다. Illumina 플랫폼은 오차율이 낮은 100~300 bp의 short-read 데이터를 생성하는 반면, Pacific Biosciences나 Nanopore 플랫폼은 15 kb 이상의 long-read 데이터를 생성하지만 이에 비례하여 오차율이 높은 특성이 있다. 따라서 시퀀싱 데이터의 특성에 맞는 적절한 조립 도구(assembler)를 선택하는 것이 중요하다.
일반적으로 long-reads는 조립 과정을 단순화하고 보다 정확한 게놈 분석을 제공하기 때문에 short-reads 보다는 long-reads가 유전체 조립에 유리하다. 또한 short-reads 데이터는 반복 시퀀스 영역(repeat sequence regions)에서 조립이 실제로 생성되는지 여부를 판단하는 것이 불가능할 수 있으며, 이로 인해 거짓 양성의 중복율이 높아지고, 불완전한 게놈 분석을 할 수 있다.

2.2. 유전체 조립(Genome Assembly)
선도 유전체 조립(de novo assembly)은 참조 유전체(reference genome)가 없는 종에 대한 유전체 시퀀스를 얻는 과정을 말한다. NGS를 통해 생성된 서열 분석 데이터를 이용하는 de novo assembly 방식은 overlap-layout-consensus 방식과 de Bruijn graph 방식의 알고리즘이 주로 사용되고 있다(Miller et al., 2010). 이러한 방법은 서로 다른 분석 서열 사이에서 가장 긴 중복된 지역을 검색하는 데 사용되며, 이러한 중복 지역은 점차적으로 결합되어 콘티그(contigs)를 형성한다.
De Bruijn graph는 reads가 아닌 k-mer를 기반으로 하기 때문에 높은 중복성은 노드 수에 영향을 주지 않고 그래프로 처리한다. 각 반복은 그래프에서 한 번만 제시되며, 다른 시작점과 끝점에 대한 명시적 링크가 있다. 이 접근법의 장점은 반복이 쉽게 인식되는 반면 어셈블리 결과의 정확성은 read insert size 및 k-mer 크기 설정의 영향을 받으므로 최적의 파라미터 설정이 필요하다. De Bruijn graph를 사용하는 assembler는 SOAPdenovo2 (Luo et al., 2012), ALLPATHS (Butler et al., 2008), AbySS (Simpson et al., 2009),Velvet (Zerbino and Birney, 2008) 등이 사용되고 대부분 short-reads 데이터를 이용한 조립 분석에 사용된다.
Overlap-layout-consensus 방법에서는 reads 간의 관계를 중첩 그래프(overlap-graph)로 나타낼 수 있다. 각각의 노드는 각각의 reads를 나타내고 각각의 reads가 겹치면 끝에서 두 개의 노드를 연결한다. 알고리즘은 모든 노드를 포함하는 그래프를 통해 Hamilton path를 결정한다. Overlap-layout-consensus 방법에 기반한 assembler로는 Celera assembler (Myers et al., 2000), CAP3 (Huang and Madan, 1999), MaSuRCA (Zimin et al., 2013), Canu (Koren et al., 2017) 등이 사용되고 long-reads 데이터를 분석하기에 적합하다. 최근에는 long-reads 데이터를 이용하여 분석 가능한 여러 새로운 알고리즘을 기반으로 FALCON-Unzip (Chin et al., 2016), Flye (Kolmogorov et al., 2019), Hifiasm (Cheng et al., 2021) 등이 개발되어 사용되고 있다.
2.3. Long-read sequencing을 이용한 유전체 분석
Long-read sequencing 또는 3rd generation sequencing은 short-reads 서열 분석보다 많은 장점을 제공한다. Short-read 시퀀싱은 상대적으로 저렴한 비용으로 효율적이고 정확한 데이터를 대량으로 얻을 수 있으며 다양한 분석 도구 및 파이프 라인을 지원한다. 그러나 거대한 유전체를 짧게 증폭된 조각으로 나눈 후 배열하는 것은 유전체 조립 과정을 복잡하게 만든다. 따라서 long-read 서열 분석은 de novo assembly, mapping의 정확성, 전사 아이소폼(transcript isoform) 확인과 구조적 변이의 확인을 향상시킬 수 있다(Amarasinghe et al., 2020). 최근 long-read sequencing의 정확성, 처리량 및 비용 절감의 지속적인 발전과 함께 long-read sequencing은 점차 모델 및 비모델 생물의 유전체 해독에 광범위하게 이용되고 있다(Koren and Phillippy, 2015; Liang et al., 2020; Logsdon et al., 2020; Nath et al., 2021). 현재 long-read sequencing 기술은 Pacific Biosciences (PacBio)의 Single-Molecule Real-Time (SMRT) sequencing과 Oxford Nanopore Technologies (ONT)의 nanopore sequencing이 있으며, SMRT 기술과 nanopore 시퀀싱 기술은 2011년과 2014년에 각각 상용화 되었고, 이후 점점 응용 분야가 확대되고 있다.
SMRT sequencer (RSII, Sequel, Sequel IIe)는 SMRT cell 내에 고정되어 있는 polymerase 효소에 의해 특정 뉴클레오티드가 합성될 때 각각의 형광을 실시간으로 감지한 동영상을 통하여 염기서열 분석을 진행한다. SMRT 염기서열 분석에서 읽는 길이는 poly- merase의 수명에 의해 제한되며, 2021년 read 길이를 평균 30 kb의 read 길이까지 가능하다.
Nanopore sequencer (MinION, GridION, PromethION)는 단일 가닥 핵산이 생물학적 나노포어를 통과할 때 이온 전류 변동을 측정한다. 즉, 서로 다른 뉴클레오타이드는 nanopore 내에서 서로 다른 저항을 부여하므로, 염기 순서는 전류변화의 특정한 패턴으로부터 추론될 수 있다. Nanopore sequencing은 현재 최대 2.3 Mb까지 가장 긴 서열 분석 길이를 제공하였으며, 일반적으로 10~30 kb의 서열 분석이 일반적이다.
2.4. 전사체 조립(Transcriptome Assembly)
전사체(mRNA)의 조립을 위해서는 de Bruijn graph method 기반의 Trinity (Henschel et al., 2012)와 Oases (Schulz et al., 2012)가 유용하게 이용되고 있다. Trinity assembler는 Inchworm, Chrysalis와 Butterfly의 3개의 모듈로 구성되어 있으며, assembly module인 Inchworm은 먼저 RNA 서열 분석 reads의 k-mer graph를 형성한 후 그리드 방식으로 그래프를 교차시켜 contigs를 생성한다. 클러스터링 모듈인 Chrysalis는 대체 접합(alternative splicing)이나 유사 유전자 부위에서 발생하는 contigs들을 결합해 클러스터를 구성하고, 각 클러스터 별로 de Bruijn graph를 구축한다. 마지막으로 Butterfly는 de Bruijn graph를 최적화한 다음 최적화된 그래프를 통하여 transcript를 생성한다. Oases는 velvet과 유사하게 다양한 k-mer (single-end의 경우 21~35, paired-end의 경우 17~40)에 의해 분석된 결과를 topological analysis와 유사한 방식으로 merge하여 결과를 도출 한다. Topology를 방해하는 noise를 제거하기 위해 dynamic correction method가 내장되어 있다.
2.5. Long-read sequencing을 이용한 전사체 분석
대체 접합(alternative splicing)은 진핵생물에서 유전자 발현의 복잡성을 증가시키는 주요 메커니즘이다. 실질적으로, 진핵생물의 모든 다중 엑손 유전자들은 조직 간의 또는 개체 간의 대체 접합을 갖는다. 그러나 short-reads 데이터를 이용하여서는 발현되는 isoforms을 완전히 assembly하거나 정확하게 정량화 할 수 없다. Long-read transcripts sequencing은 mRNA 서열을 assembly 과정을 거치는 것이 아니라 mRNA의 5'-부터 poly-A tail까지 단일분자의 전체 서열을 sequencing 함으로써 해결책을 제공한다(Gonzalez-Garay, 2016; Kuo et al., 2020). 단일분자 mRNA의 염기서열 분석은 다양한 아이소폼의 정보뿐만 아니라 RNA 변형 또는 poly-A tail 길이와 같은 또 다른 특성의 정보를 제공 한다.
Long-read transcriptomes의 분석을 위한 pipeline으로는 SMRT 데이터를 분석할 수 있는 PacBio의 IsoSeq3 (https://github.com/ PacificBiosciences/IsoSeq), SQANTI (Tardaguila et al., 2018) 외에 LIQA (Hu et al., 2021), IDP (Fu et al., 2018), Mandalorian (Byrne et al., 2017), Aeron (Rautiainen et al., 2020), TALON (Wyman et al., 2020)과 같은 많은 분석 도구가 개발되고 있다.
2.6. 게놈 주석(Genome Annotation)
게놈 주석은 게놈 시퀀스에 관련된 모든 특징을 식별하고 첨부하는 과정을 말하며 구조 주석(structural annotation)과 기능 주석(functional annotation)으로 나뉜다. 구조 주석은 조립된 염기서열 데이터를 이용해 이들 유전자를 구성하는 exons, introns, UTRs 등 유전자 위치와 구조를 식별하는 과정이다. 기능 주석은 유전자의 생화학적 및 대사 활동, 세포 및 생리적 기능을 식별하는 과정을 말한다.
2.6.1. 구조 주석(Structural Annotation)
(1) Repeat Identification
Repeat은 유전체 서열 전체에서 발견되며 2 bp (simple repeats)에서 10 kb (interspersed repeats)까지 다양하다. 유전자 예측에 앞서 repeat은 잘못된 상동성(false homology) 예측을 발생하므로 low-complexity regions과 transposable elements를 포함한 repeat elements를 가리는 것이 중요하다. 이의 과정을 위해서 Repeat- Masker (http://repeatmasker.org) 프로그램이 널리 이용되고 있다.
(2) Ab Initio gene 구조 예측
유전자 구조를 예측하기 위해 ab Initio 유전자 예측은 유전자 구조가 잘 알려진 Caenorhabditis elegans, Drosophila melanogaster, humans 등의 유전자 모델에 대한 구조적 정보로부터 얻은 미리 계산된 파라미터 값을 사용한다. 새로운 게놈이 미리 계산된 매개 변수를 이용할 수 있는 유전자 모델과 밀접한 관련이 있지 않으면 유전자 예측은 연구 중인 유전체에 대한 훈련이 필요하다. Augustus (Stanke et al., 2006), SNAP (Korf, 2004), GlimmerHMM (Majoros et al., 2004), GeneMark-ES (Lomsadze et al., 2005)는 ab initio 도구의 대표적인 예이다.
(3) Evidence-Driven Prediction of Gene Structure
대부분의 ab initio gene prediction tools은 CDS 구조만 찾을 수 있을 뿐 UTRs이나 alternatively spliced transcripts는 찾을 수 없다. 따라서 외부 증거를 바탕으로 유전자 구조를 예측하는 여러 방법이 사용되고 있다. 증거기반 방식은 expressed sequence tags (EST), 단백질 염기서열, RNA-seq 데이터 등을 유전체 어셈블리에 정렬해 얻은 결과를 외부 증거로 활용한다. 이 방법은 증거 정렬을 기반으로 유전자 예측 프로그램의 결과와 통합하여 유전자 예측의 품질을 향상시킨다. MAKER (http://www.yandell-lab.org/software/ maker.html)는 대표적인 증거 중심 구조 예측 도구의 하나로서, 여러 소프트웨어 툴을 하나로 묶는 분석 파이프라인 도구이다. 이 도구에는 RepeatMasker, Exonerate, SNAP, Augustus, Blast 등이 포함되어 있다.
(4) Evidence-Based Consensus Gene Prediction
게놈에서 유전자 구조를 도출하기 위해 다양한 유전자 예측 방법과 도구를 사용할 때 이러한 결과를 결합해 하나의 일치된 유전자 구조를 얻어내는 것이 필수적이다. 컨센서스 유전자 예측 도구에는 EvidenceModeler (EVM) (https://evidencemodeler.github.io) (Haas et al., 2008), GLEAN (https://sourceforge.net/projects/glean-gene) (Elsik et al., 2007), Evigan (http://www.seas.upenn.edu/∼strctlrn /evigan/evigan.html) (Liu et al., 2008) 등이 있다. 이들 도구는 유전자 증거로 발생하는 오류의 종류와 빈도를 추정한 뒤 이러한 오류를 최소화하는 증거를 복합적으로 선택해 consensus gene structure를 추출한다(Liu et al., 2008).
2.6.2. 기능 주석(Functional Annotation)
기능 주석은 유전자나 단백질 서열에 생물학적 정보를 붙이는 과정으로, 기능 주석은 크게 Blast-based homology search과 ontology-based GO term mapping이 있다.
(1) Homology Search
유전자 기능을 조사하거나 관련 서열 간 진화적 연관성을 예측하기 위해 새롭게 조립된 서열은 알려진 기능이 있는 유전자 서열과 비교해 상동성이 높은 서열을 찾는다. 상동 검색을 위한 도구로는 Blast (https://blast.ncbi.nlm.nih.gov) (Johnson et al., 2008), InterProScan (Jones et al., 2014), Trinotate (Bryant et al., 2017)이 있다.
(2) GO Term Mapping
Blast 검색을 통해 얻은 정보와 관련된 GO terms (Gene Ontology term)을 검색하는 과정이다(Gene ontology consortium, 2015). Gene ontology는 gene-related terms와 유전자 간의 관계에 대한 정보를 저장한다. Gene ontology는 크게 biological process ontology, molecular function ontology, cellular component ontology의 3가지로 분류된다.
(3) KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analysis
KEGG pathway는 유전자 산물의 대사 경로에 관여하는 효소를 분석하고 유전자 산물 간의 상호작용을 예측할 수 있다(Kanehisa et al., 2007). KEGG 경로는 수많은 분자 간의 상호작용을 나타내고 대사 활동을 보여주는 네트워크 도표이다.
3. 양식산업의 유전체 연구
3.1. 수산양식종의 유전체 연구
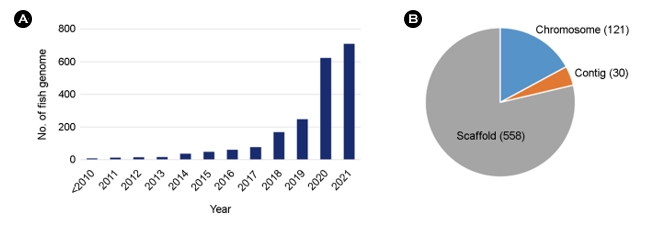
2005년경에 시작된 NGS 기술의 개발과 발전에 따라 700개 이상의 어류 유전체가 분석 공개되어 사용할 수 있게 되었다. 2021년 9월 기준으로 미국 국립생명공학정보국(NCBI)에는 709종의 어류의 유전체 정보가 공개되어 있다(Fig. 2). 이들과 다른 다양한 유전체 자원들은 비교 유전체학, 진화, 환경 적응과 같은 기초과학뿐만 아니라 양식과 수산업에서의 활용 연구를 촉진하고 있다.
유전체 서열이 밝혀진 주요 양식종(Table 1)은 메기(Catfish) (Liu et al., 2016), 대서양 연어(Atlantic salmon) (Lien et al., 2016), 무지개 송어(Rainbow trout) (Berthelot et al., 2014), 틸라피아(Tilapia) (Brawand et al., 2014), 줄무늬농어(Striped bass), 태평양 굴(Pacific oyster) (Zhang et al., 2012), 흰다리새우(Pacific white shrimp), 블루길(Bluegill sunfish) 등 다양한 생물종들이 연구되고 있다. 이들 유전체 연구는 국제적 협력을 통하여 성과가 이루어 졌다. 예를 들어 태평양 굴 게놈 프로젝트는 중국과 미국, 대서양 연어 프로젝트는 노르웨이, 캐나다, 칠레, 무지개 송어 유전체 해독은 프랑스 과학자들이 주도했고 현재는 주로 미국과 노르웨이의 공동 연구가 지속적으로 이루어지고 있으며, 태평양 흰다리새우 유전체 프로젝트는 중국 주도로 참조 유전체가 해독되었다.
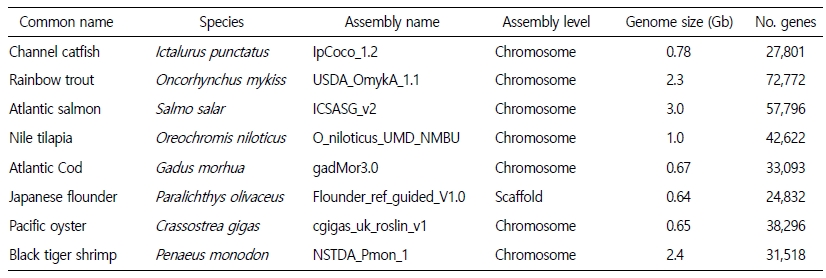
수산양식종의 유전체 시퀀싱 발전에는 Illumina와 Pacbio 플렛폼이 가장 크게 기여했다. Illumina sequencing은 비교적 저렴한 비용으로 정확하지만 짧은 서열을 대량으로 생성하는 반면, Pacbio sequencing은 상대적으로 높은 비용이지만 더 긴 유전자 서열을 생성하여 조립 과정의 많은 장점을 제공하였다. 최근 Pacbio sequencing 비용이 크게 감소하면서 이 기술의 사용이 증가하여 유전체 조립의 품질이 크게 향상되었다. 유전체의 조립품질은 1) Contiguity, contig의 개수와 contig 크기의 분포; 2) Connectivity, scaffolds의 개수와 scaffolds 크기의 분포; 3) Completeness, 유전체 조립 크기와 전체 유전체의 coverage; 4) Accuracy, 유전적 연계 맵핑(genetic linkage mapping), 물리적 매핑(physical mapping)과 같은 적어도 하나의 추가 방법론에 의해 검증된 정확성과 같은 크게 4가지의 평가로 이루어진다. 메기, 틸라피아, 대서양 연어, 무지개 송어 등의 유전체는 완성도가 매우 높다. 메기의 참조 게놈 시퀀스는 re-sequencing 결과 99.7%가 참조 게놈 시퀀스에 맵핑되면서 완성도가 매우 높은 것으로 평가된다. 또한 참조 게놈 서열의 99.1%는 염색체에 anchoring 되어 있고, 253,744개의 유전적으로 맵핑된 SNPs가 참조 게놈에 일치하였다(Liu et al., 2016). 대서양 연어의 참조 게놈도 고품질로 조립되었다. 이 유전체는 Sanger와 Illumina 기술로 염기서열을 분석했다. 일부 repeat 지역을 제외하고 2.97 Gb 참조 게놈 유전체가 완성되었고 유전체 서열의 82% 이상이 염색체에 맵핑되었다. 대서양 연어 유전체는 최근의 게놈 복제(genome duplication) 때문에 대부분 4배체이고, 높은 반복 서열(repeat content) (58~60%)을 가지고 있는 등 대서양 연어 유전체의 매우 복잡한 특성을 고려할 때 우수한 품질로 평가된다(Lien et al., 2016). 틸라피아 유전체 염기서열은, 최근 PacBio SMRT 시퀀싱 기술을 사용하면서 새로운 고품질 유전체를 조립할 수 있게 되었다(Conte et al., 2017). Contigs L50 길이는 3.09 Mb에 도달했고, 가장 큰 93개 contigs에는 게놈의 50%가 포함돼 있다. 게놈 시퀀스의 86.9% 이상이 염색체에 고정되어 있어 참조 게놈 시퀀스의 유용성을 향상시켰다. 국내 연구에서는 2018년도에 세발낙지(Octopus minor)의 유전체를 long-read sequencing을 통하여 조립하였다(Kim et al., 2018). 세발낙지의 유전체는 두족류 중 가장 큰 게놈 크기(5.09 Gb)를 가지고 있었다. 또한 황복(Takifugu obscurus)의 22개의 염색체를 완전 해독하였고, 분석된 유전체 서열의 90% 이상이 염색체에 고정되어 있어 유전 분석을 위한 참조 게놈 시퀀스로서 효용성이 매우 높다고 할 수 있다(Kang et al., 2020).
3.2. 수산양식종의 전사체 연구
유전체의 주석은 부분적으로는 Transcriptome 정보로 정확도를 높일 수 있다. 유전자 모델과 유전자 구조는 실험 데이터에 의해 뒷받침되어야 하고, Exon-intron 경계가 정의되어야 하며, 접합 변이(alternatively spliced isoforms)를 식별하고 그 번역된 단백질이 검증되어야 하며, 유전자의 발현과 기능을 연구할 필요가 있다. 단백질 coding 유전자 외에도 non-coding RNA를 확인하고 상호작용의 메커니즘을 이해할 필요가 있다. 다수의 EST 자원들이 주요 양식종들에서 보고되었다(Table 2). 대서양 연어의 경우 거의 82만개의 EST, 메기의 경우 35만개, 무지개 송어의 경우 80만개의 EST가 보고되었다. 이러한 EST 자원은 유전체 주석을 위하여 유용할 뿐 만 아니라, 저비용의 NGS sequencing 기술이 등장함에 따라 효율적으로 RNA-seq으로도 활용되고 있다. 중요 양식종에 대한 대규모 RNA-seq 데이터가 다양한 연구팀에서 진행되었다. 예를 들어 연어의 질병 그리고 성장과 관련된 유전자 마커를 식별하는 것(Valenzuela-Miranda et al., 2015; Xu et al., 2015a), 무지개 송어의 질병 및 조직 특이성(Marancik et al., 2015; Narum and Campbell, 2015; Salem et al., 2012), 줄무늬농어에서 생식 형질과 난자의 질에 초점을 맞췄고(Chapman et al., 2014; Reading et al., 2012; Sullivan et al., 2015), 틸라피아에서는 알칼리성 스트레스(Zhao et al., 2015), 염분 적응(Xu et al., 2015b), 저 · 고지방 식단 적응에 반응하는 유전자를 확인했다(He et al., 2015). 새우에서는 RNA-seq 분석을 통해 초기 발달과 관련된 유전자(Wei et al., 2014)와 Taura syndrome virus (TSV)에 대한 내성 관련 유전자가 보고되었다(Sookruksawong et al., 2013).
3.3. 유전자 발현 조절로서 Non-coding transcripts
수산양식종의 유전자 발현을 조절하는 것에 있어서 non-coding transcripts에 대한 연구는 주로 무지개 송어에서 microRNA와 long non-coding RNAs에 대한 연구만이 제한적으로 이루어졌다. 무지개 송어의 성적으로 성숙한 물고기와 미성숙한 물고기 사이에서 많은 수의 microRNA가 차등적으로 발현되며 이는 난자의 품질과 근육 성장 및 품질과 관련된다고 보고하였다(Juanchich et al., 2016; Mennigen et al., 2012; Mennigen et al., 2013; Mennigen and Zhang, 2016).
3.4. 유전체 기반의 기술
최근 유전체 연구에 데이터는 점점 대형화 되어감에 따라 생명정보학적 분석도 Machine learning을 적용한 분석이 발전하고 있다. 지도 학습(Supervised machine learning)은 Support Vector Machines (SVMs)과 인공신경망(ANN: Artificial Neural Network)을 포함한 훈련 및 테스트된 데이터 세트를 사용하는 전체 유전체 분석이다(D'Agaro, 2018; Mittag et al., 2012). 이들은 특정 데이터 입력 패턴을 인식하도록 훈련된 다음 결과를 예측하거나 데이터를 분류하는 데 사용할 수 있는 시스템이다. Machine learning은 패턴 인식으로 전사체나 단백체 데이터를 분류하는 데 사용된다. 유전자와 단백질의 발현 패턴은 형질이나 생물의 반응에 기여하는 가장 중요한 요인을 식별하기 위해 모델이 된다. 줄무늬농어 대한 microarray와 RNA-Seq 연구에서 233개의 난소 유전자의 변이가 난소의 생존율에 영향을 미친다는 것을 Machine learning ANNs을 통하여 분석되었다(Chapman et al., 2014; Sullivan et al., 2015). 또한 훈련된 ANNs은 배란 전에 채취한 난소 조직의 유전자 발현 프로파일을 토대로 암컷 줄무늬 농어가 가임 또는 난임 난자를 만들어내는 정확한 분류율을 80% 이상으로 예측하고 있다. 또한 SVMs은 줄무늬 농어의 난소 단백체을 모델링 하는 데 사용되었으며, 이 시스템은 정량적 tandem mass spectrometry 데이터를 기반으로 83%의 정확도로 구체적인 난소 성장 단계를 예측하였다(Reading et al., 2013). 따라서 기계 학습은 나쁜 품질의 난자를 생산할 암컷을 식별하거나 생식 상태 또는 성별을 결정하는 것과 같은 진단 도구로서의 사용 가능성을 제공할 수 있다. Machine learning의 향후 응용 프로그램에는 특정 형질을 결정짓는 중요한 SNP와 같은 유전 마커를 모델링 하는 다양한 분야에 적용될 것이다.
유전체 편집(Genome editing)은 표적이 되는 유전체 부위에서 특이적인 변화를 가능하게 한다(Nemudryi et al., 2014). 1996년 개발된 zinc finger nuclease (ZFN) 기술 이후로 게놈 편집은 TALEN (transcription activator-like effector nucleases), CRISPR/Cas9 (clus- tered regulatory interspaced short palindromic repeats)로 훨씬 정교하고 효율적인 기술로 발전해 왔다. 게놈 편집 기술은 단일 세대에 표현형의 즉각적인 개선을 도입하는 데 사용될 수 있다. 따라서 이러한 기술은 양식종의 개량을 위한 큰 가능성을 가지고 있다. 그러나 유전자변형생물(genetically modified organisms, GMO)은 특히 양식 생물과 관련하여 대중의 수용성이 낮다. 그러나 유전자 편집 기술이 기존 유전자변형 기술과 다른 점은 외래 유전자가 도입되지 않기 때문에 과학계는 규제 문제와 대중의 인식 분야에서 유전체 편집 기술의 도입에 전향적일 필요가 있다.
4. 결론
경제적으로 중요한 양식생물종은 다양한 생물군을 포함하고 있으며, 이들의 연구는 각 생물종의 고유한 생물학적 특성에 따라 연구 우선순위가 달라진다. 어류는 가장 다양한 척추동물군이지만 수산양식 되는 어류는 계통학적, 생물학적으로 유사한 특성을 많이 포함하고 있다. 수산양식생물종에 대한 유전체 연구의 적용은 우선 종별 유전체 자원 확보와 이를 활용한 유전적 기능을 이해하는 연구로 이어지고 있다. 예를 들어, 많은 연구팀이 어류의 성별 결정의 근본이 되는 유전자 조절 네트워크에 대한 연구나 성장, 질병 내성 등 중요한 형질의 유전적 근거를 이해하기 위한 연구가 이루어지고 있다. 이와 함께, 우수 형질의 경제성을 높이기 위해 형질을 개선하기 위한 구체적인 유전자 수정도 가능해질 것이다. 이는 수산양식종의 귀중한 자원으로서 보호와 이를 개발하기 위한 투자가 지속적으로 이뤄져야 할 것이다.
- References
-
1. Amarasinghe SL, Su S, Dong X, Zappia L, Ritchie ME, Gouil Q. 2020. Opportunities and challenges in long-read sequencing data analysis. Genome Biol 21: 1-16.

-
2. Berthelot C, Brunet F, Chalopin D, Juanchich A, Bernard M, Noël B, Bento P, Da Silva C, Labadie K, Alberti A, et al. 2014. The rainbow trout genome provides novel insights into evolution after whole-genome duplication in vertebrates. Nat Commun 5: 1-10.
-
3. Brawand D, Wagner CE, Li YI, Malinsky M, Keller I, Fan S, Simakov O, Ng AY, Lim ZW, Bezault E, et al. 2014. The genomic substrate for adaptive radiation in African cichlid fish. Nature 513: 375-381.
-
4. Bryant DM, Johnson K, DiTommaso T, Tickle T, Couger MB, Payzin-Dogru D, Lee TJ, Leigh ND, Kuo T-H, Davis FG, et al. 2017. A tissue-mapped axolotl de novo transcriptome enables iden- tification of limb regeneration factors. Cell Rep 18: 762-776.
-
5. Butler J, MacCallum I, Kleber M, Shlyakhter IA, Belmonte MK, Lander ES, Nusbaum C, Jaffe DB. 2008. ALLPATHS: de novo assembly of whole-genome shotgun microreads. Genome Res 18: 810-820.
-
6. Byrne A, Beaudin AE, Olsen HE, Jain M, Cole C, Palmer T, DuBois RM, Forsberg EC, Akeson M, Vollmers C. 2017. Nanopore long-read RNAseq reveals widespread transcriptional vari- ation among the surface receptors of individual B cells. Nat Commun 8: 1-11.
-
7. Chapman RW, Reading BJ, Sullivan CV. 2014. Ovary transcriptome profiling via artificial intelligence reveals a transcriptomic fingerprint predicting egg quality in striped bass, Morone saxatilis. PLoS One 9: e96818.
-
8. Cheng H, Concepcion GT, Feng X, Zhang H, Li H. 2021. Haplotype-resolved de novo assembly using phased assembly graphs with hifiasm. Nat Methods 18: 170-175.
-
9. Chin CS, Peluso P, Sedlazeck FJ, Nattestad M, Concepcion GT, Clum A, Dunn C, O'Malley R, Figueroa-Balderas R, Morales-Cruz A, et al. 2016. Phased diploid genome assembly with single-molecule real-time sequencing. Nat Methods 13: 1050-1054.
-
10. Conte MA, Gammerdinger WJ, Bartie KL, Penman DJ, Kocher TD. 2017. A high quality assembly of the Nile Tilapia (Oreochromis niloticus) genome reveals the structure of two sex deter- mination regions. BMC Genomics 18: 1-19.
-
11. D'Agaro E. 2018. Artificial intelligence used in genome analysis studies. Eurobiotech J 2: 78-88.
-
12. Elsik CG, Mackey AJ, Reese JT, Milshina NV, Roos DS, Weinstock GM. 2007. Creating a honey bee consensus gene set. Genome Biol 8: 1-8.
-
13. FAO. 2018. GLOBEFISH Highlights 4th issue 2018. A quarterly update on world seafood markets. FAO Globefish Research Programme.
-
14. Fu S, Ma Y, Yao H, Xu Z, Chen S, Song J, Au KF. 2018. IDP-denovo: de novo transcriptome assembly and isoform annotation by hybrid sequencing. Bioinformatics 34: 2168-2176.
-
15. Gene ontology consortium. 2015. Gene ontology consortium: going forward. Nucleic Acids Res 43: D1049-D1056.
-
16. Gonzalez-Garay ML. 2016. Introduction to isoform sequencing using pacific biosciences technology (Iso-Seq). Transcriptomics and gene regulation. Springer. pp 141-160.
-
17. Haas BJ, Salzberg SL, Zhu W, Pertea M, Allen JE, Orvis J, White O, Buell CR, Wortman JR. 2008. Automated eukaryotic gene structure annotation using EVidenceModeler and the Program to Assemble Spliced Alignments. Genome Biol 9: 1-22.
-
18. He AY, Ning LJ, Chen LQ, Chen YL, Xing Q, Li JM, Qiao F, Li DL, Zhang ML, Du ZY. 2015. Systemic adaptation of lipid metab- olism in response to low-and high-fat diet in Nile tilapia (Oreochromis niloticus). Physiol Rep 3: e12485.
-
19. Henschel R, Lieber M, Wu L-S, Nista PM, Haas BJ, LeDuc RD. 2012. Trinity RNA-Seq assembler performance optimization. Proceedings of the 1st Conference of the Extreme Science and Engineering Discovery Environment: Bridging from the eXtreme to the campus and beyond. ACM. New York. pp 1-8.
-
-
21. Hu Y, Fang L, Chen X, Zhong JF, Li M, Wang K. 2021. LIQA: long-read isoform quantification and analysis. Genome Biol 22: 1-21.
-
22. Johnson M, Zaretskaya I, Raytselis Y, Merezhuk Y, McGinnis S, Madden TL. 2008. NCBI BLAST: a better web interface. Nucleic Acids Res 36: W5-W9.
-
23. Jones P, Binns D, Chang HY, Fraser M, Li W, McAnulla C, McWilliam H, Maslen J, Mitchell A, Nuka G, et al. 2014. InterProScan 5: genome-scale protein function classification. Bioinformatics 30: 1236-1240.
-
24. Juanchich A, Bardou P, Rué O, Gabillard J-C, Gaspin C, Bobe J, Guiguen Y. 2016. Characterization of an extensive rainbow trout miRNA transcriptome by next generation sequencing. BMC Genomics 17: 1-12.
-
25. Kanehisa M, Araki M, Goto S, Hattori M, Hirakawa M, Itoh M, Katayama T, Kawashima S, Okuda S, Tokimatsu T, et al. 2007. KEGG for linking genomes to life and the environment. Nucleic Acids Res 36: D480-D484.
-
26. Kang S, Kim JH, Jo E, Lee SJ, Jung J, Kim BM, Lee JH, Oh TJ, Yum S, Rhee JS, et al. 2020. Chromosomal-level assembly of Takifugu obscurus (Abe, 1949) genome using third-generation DNA sequencing and Hi-C analysis. Mol Ecol Resour 20: 520-530.
-
27. Kim B-M, Kang S, Ahn D-H, Jung S-H, Rhee H, Yoo JS, Lee J-E, Lee S, Han Y-H, Ryu K-B, et al. 2018. The genome of common long-arm octopus Octopus minor. Gigascience 7: giy119.
-
28. Kolmogorov M, Yuan J, Lin Y, Pevzner PA. 2019. Assembly of long, error-prone reads using repeat graphs. Nat Biotechnol 37: 540-546.
-
29. Koren S, Phillippy AM. 2015. One chromosome, one contig: com- plete microbial genomes from long-read sequencing and assembly. Curr Opin Microbiol 23: 110-120.
-
30. Koren S, Walenz BP, Berlin K, Miller JR, Bergman NH, Phillippy AM. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res 27: 722-736.
-
-
32. Kuo RI, Cheng Y, Zhang R, Brown JW, Smith J, Archibald AL, Burt DW. 2020. Illuminating the dark side of the human transcrip- tome with long read transcript sequencing. BMC Genomics 21: 1-22.
-
33. Liang P, Saqib HSA, Ni X, Shen Y. 2020. Long-read sequencing and de novo genome assembly of marine medaka (Oryzias melastigma). BMC Genomics 21: 1-15.
-
34. Lien S, Koop BF, Sandve SR, Miller JR, Kent MP, Nome T, Hvidsten TR, Leong JS, Minkley DR, Zimin AJN, et al. 2016. The Atlantic salmon genome provides insights into rediploidization. Nature 533: 200-205.
-
35. Liu Q, Mackey AJ, Roos DS, Pereira FCN. 2008. Evigan: a hidden variable model for integrating gene evidence for eukaryotic gene prediction. Bioinformatics 24: 597-605.
-
36. Liu Z, Liu S, Yao J, Bao L, Zhang J, Li Y, Jiang C, Sun L, Wang R, Zhang Y, et al. 2016. The channel catfish genome sequence provides insights into the evolution of scale formation in teleosts. Nat Commun 7: 1-13.
-
37. Logsdon GA, Vollger MR, Eichler EE. 2020. Long-read human genome sequencing and its applications. Nat Rev Genet 21: 597-614.
-
38. Lomsadze A, Ter-Hovhannisyan V, Chernoff YO, Borodovsky M. 2005. Gene identification in novel eukaryotic genomes by self-training algorithm. Nucleic Acids Res 33: 6494-6506.
-
39. Luo R, Liu B, Xie Y, Li Z, Huang W, Yuan J, He G, Chen Y, Pan Q, Liu Y, et al. 2012. SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1: 2047-217X-1-18.
-
40. Majoros WH, Pertea M, Salzberg SL. 2004. TigrScan and Glimmer- HMM: two open source ab initio eukaryotic gene-finders. Bioinformatics 20: 2878-2879.
-
41. Marancik D, Gao G, Paneru B, Ma H, Hernandez AG, Salem M, Yao J, Palti Y, Wiens GD. 2015. Whole-body transcriptome of selectively bred, resistant-, control-, and susceptible-line rainbow trout following experimental challenge with Flavo- bacterium psychrophilum. Front Genet 5: 453.
-
42. Mennigen JA, Panserat S, Larquier M, Plagnes-Juan E, Medale F, Seiliez I, Skiba-Cassy S. 2012. Postprandial regulation of hepatic microRNAs predicted to target the insulin pathway in rainbow trout. PLoS One 7: e38604.
-
43. Mennigen JA, Skiba-Cassy S, Panserat S. 2013. Ontogenetic ex- pression of metabolic genes and microRNAs in rainbow trout alevins during the transition from the endogenous to the exogenous feeding period. J Exp Biol 216: 1597-1608.
-
44. Mennigen JA, Zhang D. 2016. MicroTrout: A comprehensive, genome-wide miRNA target prediction framework for rain- bow trout, Oncorhynchus mykiss. Comp Biochem Physiol -D: Genome Proteom 20: 19-26.
-
45. Miller JR, Koren S, Sutton G. 2010. Assembly algorithms for next-generation sequencing data. Genomics 95: 315-327.
-
46. Mittag F, Büchel F, Saad M, Jahn A, Schulte C, Bochdanovits Z, Simón-Sánchez J, Nalls MA, Keller M, Hernandez DG, et al. 2012. Use of support vector machines for disease risk pre- diction in genome-wide association studies: Concerns and opportunities. Hum Mutat 33: 1708-1718.
-
47. Myers EW, Sutton GG, Delcher AL, Dew IM, Fasulo DP, Flanigan MJ, Kravitz SA, Mobarry CM, Reinert KH, Remington KA, et al. 2000. A whole-genome assembly of Drosophila. Science 287: 2196-2204.
-
48. Narum SR, Campbell NR. 2015. Transcriptomic response to heat stress among ecologically divergent populations of redband trout. BMC Genomics 16: 1-12.
-
49. Nath S, Shaw DE, White MA. 2021. Improved contiguity of the threespine stickleback genome using long-read sequencing. G3-GENES GENOM GENET 11: jkab007.
-
50. Nemudryi A, Valetdinova K, Medvedev S, Zakian S. 2014. TALEN and CRISPR/Cas genome editing systems: tools of discovery. Acta Naturae 6.
-
51. Rautiainen M, Durai DA, Chen Y, Xin L, Low HM, Göke J, Marschall T, Schulz MH. 2020. AERON: Transcript quantification and gene-fusion detection using long reads. bioRxiv 921338.
-
52. Reading BJ, Chapman RW, Schaff JE, Scholl EH, Opperman CH, Sullivan CV. 2012. An ovary transcriptome for all matur- ational stages of the striped bass (Morone saxatilis), a highly advanced perciform fish. BMC Res Notes 5: 1-12.
-
53. Reading BJ, Williams VN, Chapman RW, Williams TI, Sullivan CV. 2013. Dynamics of the striped bass (Morone saxatilis) ovary proteome reveal a complex network of the translasome. J Proteome Res 12: 1691-1699.
-
54. Salem M, Vallejo RL, Leeds TD, Palti Y, Liu S, Sabbagh A, Rexroad III CE, Yao J. 2012. RNA-Seq identifies SNP markers for growth traits in rainbow trout. PLoS One 7: e36264.
-
55. Schulz MH, Zerbino DR, Vingron M, Birney E. 2012. Oases: robust de novo RNA-seq assembly across the dynamic range of expression levels. Bioinformatics 28: 1086-1092.
-
56. Simpson JT, Wong K, Jackman SD, Schein JE, Jones SJ, Birol I. 2009. ABySS: a parallel assembler for short read sequence data. Genome Res 19: 1117-1123.
-
57. Sookruksawong S, Sun F, Liu Z, Tassanakajon A. 2013. RNA-Seq analysis reveals genes associated with resistance to Taura syndrome virus (TSV) in the Pacific white shrimp Litopenaeus vannamei. Dev Comp Immunol 41: 523-533.
-
58. Stanke M, Keller O, Gunduz I, Hayes A, Waack S, Morgenstern B. 2006. AUGUSTUS: ab initio prediction of alternative transcripts. Nucleic Acids Res 34: W435-W439.
-
59. Star B, Nederbragt AJ, Jentoft S, Grimholt U, Malmstrøm M, Gregers TF, Rounge TB, Paulsen J, Solbakken MH, Sharma A, et al. 2011. The genome sequence of Atlantic cod reveals a unique immune system. Nature 477: 207-210.
-
60. Sullivan CV, Chapman RW, Reading BJ, Anderson PE. 2015. Transcriptomics of mRNA and egg quality in farmed fish: some recent developments and future directions. Gen Comp Endocrinol 221: 23-30.
-
61. Tardaguila M, De La Fuente L, Marti C, Pereira C, Pardo-Palacios FJ, Del Risco H, Ferrell M, Mellado M, Macchietto M, Verheggen K, et al. 2018. SQANTI: extensive characterization of long-read transcript sequences for quality control in full-length transcriptome identification and quantification. Genome Res 28: 396-411.
-
62. Valenzuela-Miranda D, Boltana S, Cabrejos ME, Yánez JM, Gallardo-Escárate C. 2015. High-throughput transcriptome analysis of ISAV-infected Atlantic salmon Salmo salar unravels divergent immune responses associated to head-kidney, liver and gills tissues. Fish Shellfish Immunol 45: 367-377.
-
63. Wei J, Zhang X, Yu Y, Huang H, Li F, Xiang J. 2014. Comparative transcriptomic characterization of the early development in Pacific white shrimp Litopenaeus vannamei. PLoS One 9: e106201.
-
64. Wyman D, Balderrama-Gutierrez G, Reese F, Jiang S, Rahmanian S, Forner S, Matheos D, Zeng W, Williams B, Trout D. 2020. A technology-agnostic long-read analysis pipeline for trans- criptome discovery and quantification. bioRxiv 672931.
-
65. Xu C, Evensen Ø, Munang'Andu HM. 2015a. De novo assembly and transcriptome analysis of Atlantic salmon macrophage/ dendritic-like TO cells following type I IFN treatment and Salmonid alphavirus subtype-3 infection. BMC Genomics 16: 1-16.
-
66. Xu Z, Gan L, Li T, Xu C, Chen K, Wang X, Qin JG, Chen L, Li E. 2015b. Transcriptome profiling and molecular pathway analysis of genes in association with salinity adaptation in Nile tilapia Oreochromis niloticus. PLoS One 10: e0136506.
-
67. Zerbino DR, Birney E. 2008. Velvet: algorithms for de novo short read assembly using de Bruijn graphs. Genome Res 18: 821-829.
-
68. Zhang G, Fang X, Guo X, Li L, Luo R, Xu F, Yang P, Zhang L, Wang X, Qi H. 2012. The oyster genome reveals stress adaptation and complexity of shell formation. Nature 490: 49-54.
-
69. Zhao Y, Wang J, Thammaratsuntorn J, Wu J, Wei J, Wang Y, Xu J, Zhao J. 2015. Comparative transcriptome analysis of Nile tilapia (Oreochromis niloticus) in response to alkalinity stress. Genet Mol 14: 17916-17926.
-
70. Zimin AV, Marçais G, Puiu D, Roberts M, Salzberg SL, Yorke JA. 2013. The MaSuRCA genome assembler. Bioinformatics 29: 2669-2677.
Copyright (C) 2025. The Korean Society of Marine Life Science