






JMLS 2021 June;6(1):38-46. Epub 2021 May 31
Copyright © 2021 by The Korean Society of Marine Life Science
Genome Survey and Microsatellite Marker Selection of Tegillarca granosa
Jinmu Kim; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Seung Jae Lee; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Euna Jo; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea; Unit of Research for Practical Application, Korea Polar Research Institute (KOPRI), Incheon 21990, Korea
Eunkyung Choi; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
Hyeon Jin Kim; Department of Aqualife Medicine, Chonnam National University, Yeosu 59626, Korea
Jung Sick Lee; Department of Aqualife Medicine, Chonnam National University, Yeosu 59626, Korea
Hyun Park; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea
- Abstract
-
꼬막 종류 중 하나인 Tegillarca granosa는 해양 이매패류로서 한국, 중국, 일본 등의 중요한 수산 자원 중 하나이다. 꼬막의 염색체 수는 2n=38로 알려져 있지만, 유전체의 크기와 유전 정보에 대해서는 아직 명확하게 알려져 있지 않다. 꼬막의 유전체 크기 예측을 위하여 NGS Illumina HiSeq 플랫폼을 이용하여 얻은 짧은 DNA 서열 정보를 통하여 in silico 분석으로 유전체 크기를 분석하였다. 그 결과 꼬막의 유전체 크기는 770.61 Mb로 예측되었다. 이후 MaSuRCA assembler를 통하여 드래프트 게놈 조립 작업을 수행하고, QDD pipeline을 이용하여 SSR (simple sequence repeats) 분석을 수행하였다. 꼬막의 유전체로부터 43,944개의 SSR을 발굴하였으며, 다이-뉴클레오타이드(di-nucleotide) 69.51%, 트라이-뉴클레오타이드(tri-nucleotide) 16.68%, 테트라-뉴클레오타이드(tetra-nucleotide) 12.96%, 펜타-뉴클레오타이드(penta-nucleotide) 0.82% 그리고 헥사-뉴클레오타이드(hexa-nucleotide) 0.03%로 구성되었다. 이후 꼬막의 유전적 다양성 연구에 활용할 수 있는 100개의 마이크로새틀라이트 마커의 프라이머 세트를 선별하였다. 앞으로 이번 연구를 통해서, 꼬막의 집단유전학적 연구와 유전적 다양성을 규명하는데 도움이 될 것이며, 나아가 동종들 간의 원산지 분류를 알아낼 수 있을 것이다.
The blood clam, Tegillarca granosa, is economically important in marine bivalve and is used in fisheries industry among western Pacific Ocean Coasts especially in Korea, China, and Japan. The number of chromosomes in the blood clam is known as 2n=38, but the genome size and genetic information of the genome are not still clear. In order to predict the genomic size of the T. granosa, the in-silico analysis analysed the genomic size using short DNA sequence information obtained using the NGS Illumina HiSeq platform. As a result, the genomic size of T. granosa was estimated to be 770.61 Mb. Subsequently, a draft genome assembly was performed through the MaSuRCA assembler, and a simple sequence repeat (SSR) analysis was done by using the QDD pipeline. 43,944 SSRs were detected from the genome of T. granosa and 69.51% di-nucleotide, 16.68% trinucleotide, 12.96% tetra-nucleotide, 0.82% penta-nucleotide, and 0.03% hexa-nucleotide were consisted. 100 primer sets that could be used for genetic diversity studies were selected. In the future, this study will help identify the genetic diversity of T. granosa and population genetic studies, and further identify the classification of origin between homogenous groups.
Keywords: Tegillarca granosa(꼬막) Illumina HiSeq platform(일루미나 하이식 플랫폼) MaSuRCA assembler SSR(마이크로새틀라이트) Homogeneous(동종)
Correspondence to: Hyun Park; Division of Biotechnology, College of Life Sciences and Biotechnology, Korea University, Seoul 02841, Korea; E-mail : hpark@korea.ac.kr
- Received
- 7 May 2021;
- Revised
- 7 May 2021;
- Accepted
- 17 May 2021.
This is an Open Access article distributed under the terms of the Creative Commons Attribution Non-Commercial License (http://creativecommons.org/licenses/by-nc/3.0/) which permits unrestricted non-commercial use, distribution, and reproduction in any medium, provided the original work is properly cited.
Language: Korean/English,
Full Text:

서 론
꼬막은 연체동물문에서 이매패강에 속하는 종이다. 이 종은 주로 한국, 중국, 태국, 필리핀, 말레이시아를 따라 서태평양 연안의 중서부 지역에 주로 서식하며(Liu et al., 2017), 과거부터 현재까지 한국, 중국, 일본의 서태평양 해안을 중심으로 수산업에 있어서 매우 의미 있는 해양식품의 자원이었다(Liu et al., 2017). 예를 들어, 2010년에는 중국 갯벌 조개류 생산량의 30%에 해당되는 약 13만 톤의 꼬막이 양식되었다(Dong-li, 2011).
따라서, 수산업, 집단 유전학, 성별격리 등을 위한 Tegillarca granosa의 유전적 영역을 해결하기 위한 많은 유전학적 연구가 진행이 되고 있다. 굴, 홍합, 가리비와 같은 유전적 정보와 전사체의 정보가 양식 산업의 중요한 원천이지만, 돌조개과에 있어서는 여전히 제한적이다(Bai et al., 2019). 게다가 이매패류를 포함한 연체동물은 유전적 다양성이 높은 것으로 나타나, 수산업에 대한 집단 유전체학과 성별 분리를 해결하기에 어렵게 만든다(Penaloza et al., 2014).
본 연구에서는 꼬막의 유전체 크기를 예측하고 드래프트 게놈 어셈블리를 통하여 마이크로새틀라이트 DNA 마커를 발굴하여 집단 유전체 연구와 종간 다양성 연구의 유전자 마커를 제시하고자 한다.
재료 및 방법
1. DNA 추출과 라이브러리 제작 및 게놈 시퀀싱
T. granosa의 샘플은 2019년 9월 대한민국 순천 벌교에서 포획되었다. 꼬막의 게놈 DNA (genomic DNA)는 근육 조직에서 추출했으며 페놀: 클로로포름: 이소아밀-알코올 방법을 이용하여 추출하게 되었다. DNA의 품질과 정량분석을 위해서 BioAnalyzer (Agilent Technologies, CA, USA)와 Qubit fluorometer (Invitrogen, Life Technologies, CA, USA)을 이용하여 확인하였다.
게놈 DNA는 ultrasonicator (Covaris, USA)를 사용하여 무작위로 350 bp 조각으로 절단하였고 paired-end library를 Illumina 제조사의 실험 방법에 따라 제작하였다. DNA 서열분석은 Illumina HiSeq 2000 (Liu et al., 2011)을 이용하여 수행하였다.
2. K-mer 분석, 게놈 조립과 마이크로새틀라이트 선별
생산된 염기서열 데이터는 어댑터, 폴리(N) 시퀀스, 낮은 품질의 리드들을 필터링하여 제거한 후, Jellyfish (Marcais and Kingsford, 2011) 외에 GenomeScope (Vurture et al., 2017)을 사용하여 K-mer 17, 19, 25를 값으로 이용하여 T. granosa의 게놈 크기를 예측했다. 그 중에서 19 K-mer를 최종 게놈 크기로 예측했다. 드래프트 유전체 서열은 MaSuRCA (Maryland Super Read Cabog Assembler) (Zimin et al., 2013)을 이용하여 조립하였으며, 이는 de Bruijn graphs 방법과 겹침 기반 조립 전략의 유연성을 기반으로 한다. 콘티그(contig)와 스캐폴드(scaffold) 수준 어셈블리 통계를 계산하기 위해 assemblathon_stats.pl script (https://github.com/ucdavis-bioinformatics/assemblathon2-analysis)을 사용했다.
꼬막의 드래프트 유전체를 활용하여 마이크로새틀라이트를 분석하기 위해 QDD 버전 3.1.2 파이프라인(Meglecz et al., 2014)을 사용하여 게놈 내 마이크로새틀라이트 반복단위를 확인했다. 파이프라인에서 설정된 한계로는 모노뉴클레오티드(mononucleotides) 반복 횟수에서 핵사뉴클레오티드(hexa-nucleotide) 반복 횟수이다. 기본 파라미터로 진행된 세 가지 QDD 파이프라인 단계를 사용하였다. 세 가지 QDD 단계로는 -config 1(1단계), -make_cons 0(2단계), -config 1(3단계) 옵션이 추가되었다.
프라이머 쌍을 선별하기 위해서 예측 PCR product 크기로는 120~150 bp와 160~180 bp, 프라이머 길이로는 20~22 bp, 프라이머 용융 온도(Tm)로는 59~60℃, 다이뉴클레오티드 모티프(di-nucleotide motifs) 선택 등의 기준에 따라 Primer3 소프트웨어(Rozen and Skaletsky, 2000)을 사용해서 선정했다.
결 과
1. 게놈 염기서열 분석 및 K-mer 분석을 이용한 게놈 크기 예측
꼬막의 DNA 염기서열은 Illumina HiSeq 2000 장비를 이용하여 분석하였다. 분석된 꼬막의 DNA 염기서열 데이터 중 전체 리드로는 350,031,048가 확인이 되었고 모두 87.86 Gb의 데이터가 생산되었다. Seqtk (https://github.com/lh3/seqtk)를 이용해서 Q20, Q30 그리고 GC 함량이 총 76.2%, 73.0%, 그리고 55.4%인 것으로 확인하였다(Table 1).
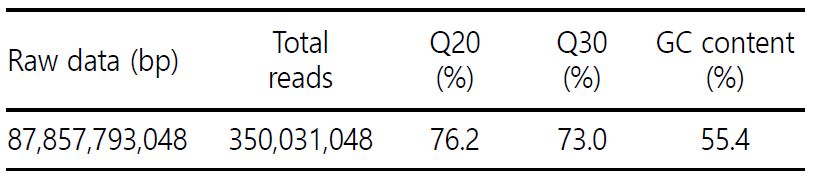
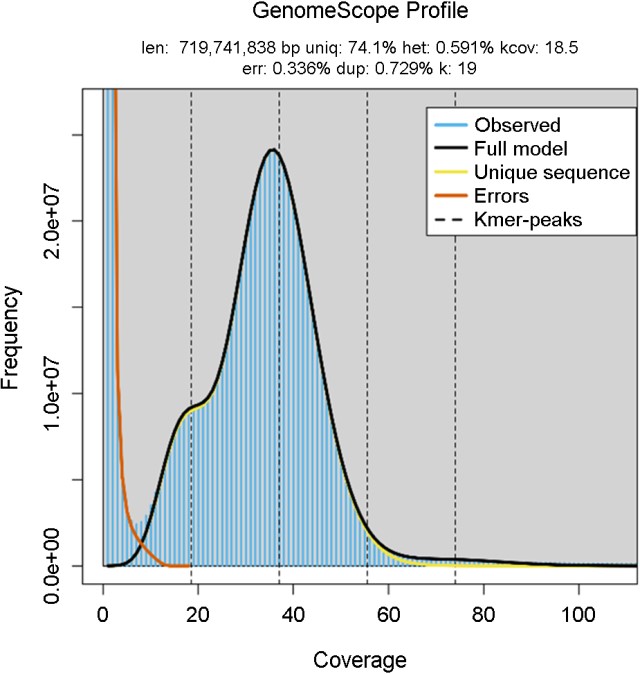
T. granosa 게놈의 정확한 크기는 보고되지 않았지만, 핵형과 염색체 수(2n=38)는 T. granosa (Lu et al., 2008)에 대해서 보고되었다. 따라서, 게놈의 크기를 예측하기 위해 in silico 분석 방법 중 하나인 K-mer 분석을 진행했고, 그 결과, K-mer 19를 이용하여 최종적으로 추정되었다. 게놈의 크기는 719,741,838 bp로 확인되었고, 이형성 비율(heterozygosity rate), 중복성 비율(duplication rate) 및 오차율(error rate)은 0.591%, 0.729%, 그리고 0.336%로 확인하였다(Fig. 1).
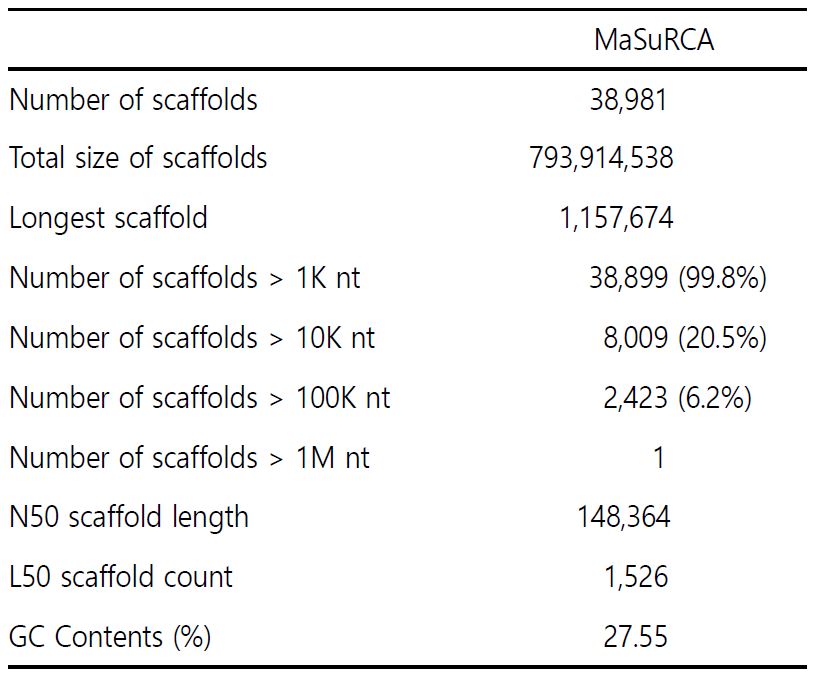
2. 게놈 조립 및 향후 연구를 위한 마이크로새틀라이트 선별
게놈 조립 도구인 MaSuRCA (Zimin et al., 2013)에 의해 꼬막의 게놈을 조립하였으며, 그 결과, 스캐폴드(scaffold)의 총 수는 38,981개이고 가장 긴 스캐폴드는 1,157,674 bp이며 스캐폴드의 총 크기로는 793,914,538 bp이다. 또한, N50 스캐폴드 길이와 GC 함량은 148,364 bp와 27.55%로 확인하였다. 이로써, T. granosa 게놈 어셈블리의 전체 크기는 770.66 Mb (Table 2)로 확인하였다.
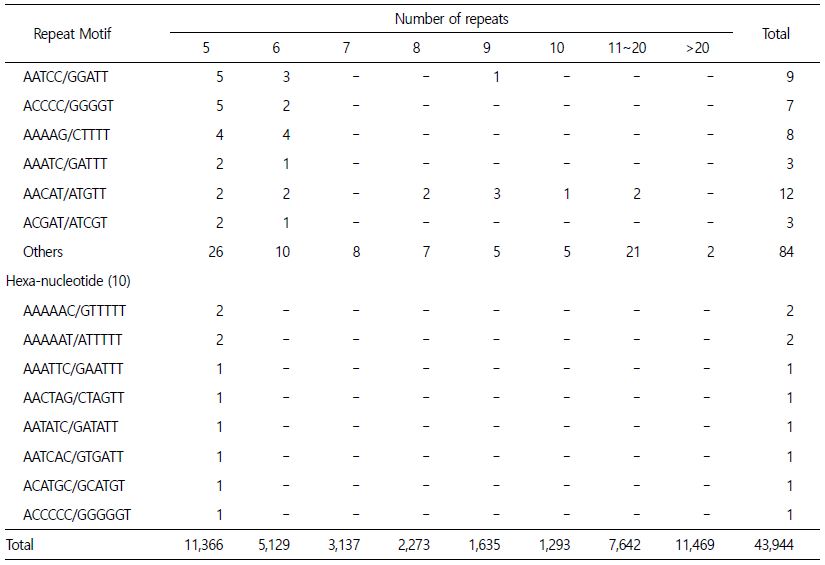
MaSuRCA에 의해 조립된 드래프트 게놈을 통하여 마이크로새틀라이트를 분석하는데 사용이 되었으며, 유전적 영역을 식별했고 QDD 버전 3.1.2 (Meglecz et al., 2014)을 사용하여 필터링을 통해 총 43,944개의 마이크로새틀라이트를 확인하였다. 검출된 전체 마이크로새틀라이트는 다이-뉴클레오타이드(di-nucleotide) 69.51% (30,548), 트라이-뉴클레오타이드(tri-nucleotide) 16.68% (7,332), 테트라-뉴클레오타이드(tetra-nucleotide) 12.96% (5,694), 펜타-뉴클레오타이드(penta-nucleotide) 0.82% (360) 그리고 헥사-뉴클레오타이드(hexa-nucleotide) 0.03% (10)이다. 다이-뉴클레오타이드 중 가장 빈번한 모티브는 AT/AT (47.62%)였고, 그 다음이 AC/GT (27.93%)였다. 트라이-뉴클레오타이드 중 가장 빈번한 모티브는 AAT/ATT (70.11%)였으며, AAC/GTT (11.82%)가 그 뒤를 이었다. 테트라, 펜타, 헥사-뉴클레오타이드 중에서 AAAT/ATTT (73.56%), AAAAT/ATTTT (44.17%), AAAAAC/GTTTTT (20%), AAAAAAT/ATTTTT (20%)가 가장 풍부한 모티브였다(Table 3).

마이크로새틀라이트를 이용한 꼬막의 기존 연구에 의하면, 중국에 있는 개체에서 나타나는 마이크로새틀라이트의 loci가 말레이시아에 사는 꼬막의 개체에서는 나타나지 않았다는 것을 실험을 통해 확인하였다(Wang et al., 2013). 아직까지 정확한 원인은 밝혀지지 않았지만, 아주 긴 시간에 걸쳐 진화가 이루어지게 되면서 어떤 중요한 유전적 분화가 말레이시아와 중국 꼬막 개체 사이에서 이루어졌을 것이라고 추정한다(Wang et al., 2013). 따라서, 꼬막의 집단 유전학 연구 등을 위한 정확한 마이크로새틀라이트 마커를 선별하기 위해서 차세대 염기서열 분석을 이용해서 더 정확하고 명확하게 이루어져야 한다.
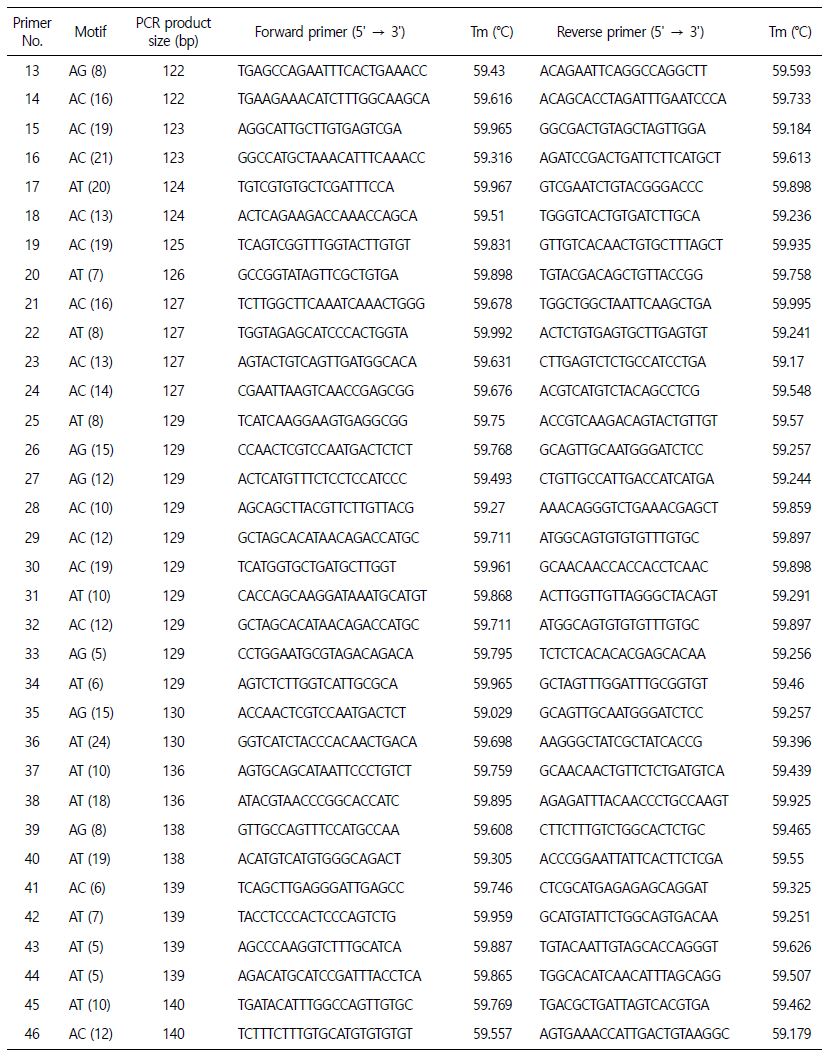
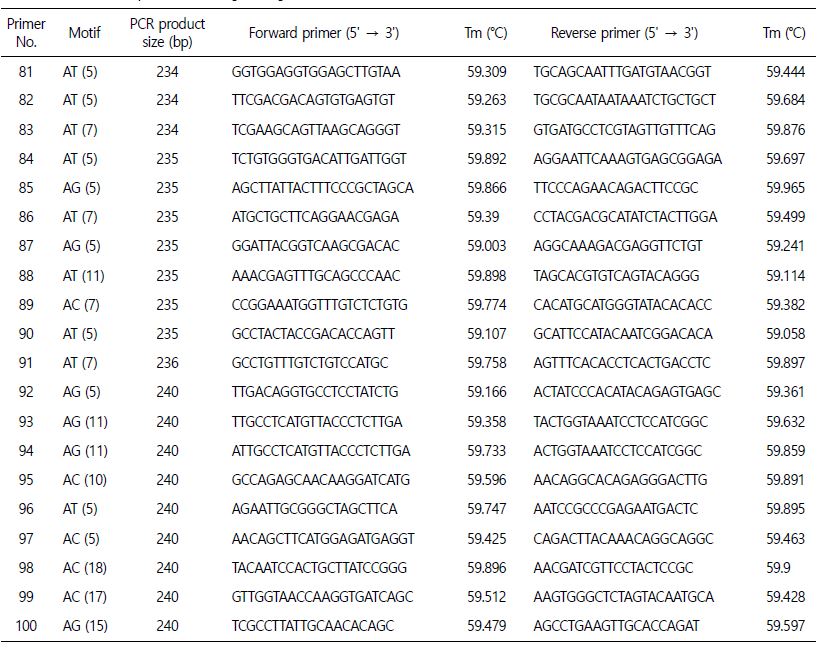
추후, 꼬막의 집단 유전학 연구 및 동종들 간의 원산지 분류를 위해 우리는 100쌍의 마이크로새틀라이트 마커를 Primer 3 소프트웨어를(Table 4) 사용하여 선별하였다. 선별한 마이크로새틀라이트 마커는 사용적합성을 검정하기 위해서는 추가적으로 후속 검증과 연구가 필요하다.


고 찰
T. granosa는 특히 한국, 중국, 일본의 서태평양 해안의 중요한 수산 자원이다(Liu et al., 2017). 꼬막이 중요한 수산 자원이기에 질병 또는 양식업에 있어서 도움이 될 수 있는 유전적 영역을 알아내려고 많은 연구와 노력들이 행해지고 있다. 물론, 잔류성 유기 오염 물질(Persistent Organic Pollutants, POPs)과 면역반응에 관한 연구(Su et al., 2017), DCMU와 같은 살충제 농도에 따른 연구(Mohamat-Yusuff et al., 2020) 등 꼬막에 관한 수많은 연구들이 이루어지고 있지만, 이러한 문제점들을 좀 더 쉽게 접근하기 위해서, 전체 게놈을 알아내는 것이 더 도움이 될 수 있다.
꼬막의 전체 게놈을 알아내기 전에, 우리는 K-mer 분석을 이용하여 게놈의 크기를 719,741,838 bp로 추정을 했으며, 중복성 비율, 이형성 비율 및 오차율은 0.729%, 0.591% 그리고 0.336%로 확인되었다(Fig. 1). T. granosa의 게놈 어셈블리 총 크기는 770.66 Mb였으며, 수집된 게놈 데이터는 QDD 버전 3.1.2 (Meglecz et al., 2014)에 의해 마이크로새틀라이트를 분석하였다. 꼬막의 마이크로새틀라이트는 총 43,944개로 확인하였다. 마이크로새틀라이트 같은 경우 진핵 유전체의 상당한 부분을 차지하고 있고, 두 가지 측면에서 모두 코딩 유전자 시퀀스를 능가할 만큼 매우 다형적이다(Katti et al., 2001). 이러한 이유 때문에, 마이크로새틀라이트의 높은 돌연변이 비율은 진화 연구에 있어서 중요하고, 모집단 유전자형 및 마커 지원 사육과 같은 매우 폭넓은 응용 분야에서도 유익하고 유용하다(Mohamat-Yusuff et al., 2020). 또한, 마이크로새틀라이트 분석을 통해 중요한 시퀀스의 특성에 대해서 더 나은 이해를 제공하고 게놈 조직에서 마이크로새틀라이트의 역할에 대한 연구를 촉진한다(Manee et al., 2020).
추가적으로 집단 유전학을 연구하기 위해, Primer 3 소프트웨어를(Rozen and Skaletsky, 2000) 이용하여 100쌍의 마이크로새틀라이트 마커를 선별하게 되었다(Table 4). 이를 통해, 유전학 및 진화 연구를 위한 풍부한 마커의 분리 및 개발을 포함하여 앞으로 많은 수산업 분야에 유용하게 도움이 될 것이다. 특히, 가장 빈번하게 일어나는 반복을 밝혀내는 것은 한 종 내의 개체를 구별하거나 심지어 이종 간에도 유용한 라이브러리 기반의 마커 선택에 있어서 필수적인 출발점을 제공하게 될 것이다(Manee et al., 2020). 이러한 데이터 기반이 갖춰지게 된다면, 이 종의 수산업 개발과 보존 계획을 지원하는데 있어서 의미 있을 것이다. 게다가, 마이크로새틀라이트는 그 종의 지리적 분포 전반에 걸친 추가적인 유전적 개체 연구 말고도, 양식활동이나 수산업 분야에 필요한 다른 유전자 분석을 가능하게 할 것이다(Fernández-Pérez et al., 2019). 앞으로의 후속연구를 위해서, 마이크로새틀라이트 마커의 사용적합성을 검정하기 위해서는 추가적인 연구와 검증이 필요할 것이다.
- References
-
1. Bai CM, Xin LS, Rosani U, Wu B, Wang QC, Duan XK, Liu ZH, Wang CM. 2019. "Chromosomal-level assembly of the blood clam, Scapharca (Anadara) broughtonii, using long sequence reads and Hi-C." Gigascience 8.


-
2. Dong-li Z. 2011. Analysis of genetic variation in the fast growth families of Tegillarca granosa. Journal of Fisheries of China 35: 350-357
-
3. Fernández-Pérez J, Nantón A, Arias-Pérez A, Insua A, Méndez J. 2019. "Fifteen novel microsatellite loci, developed using next-generation sequencing, reveal the lack of genetic structure in Donax vittatus from Iberian Peninsula." Estuarine, Coastal and Shelf Science 217: 218-225.
-
4. Katti MV, Ranjekar PK, Gupta VS. 2001. "Differential Distribution of Simple Sequence Repeats in Eukaryotic Genome Sequences." Molecular Biology and Evolution 18: 1161-1167.
-
5. Liu B, Teng S, Shao Y, Chai X, Xiao G, Fang J, Zhang J, Wang C. 2017. "A Genetic Linkage Map of Blood Clam (Tegillarca granosa) Based on Simple Sequence Repeat and Amplified Fragment Length Polymorphism Markers." Journal of Shellfish Research 36: 31-40.
-
6. Liu L, Hu N, Wang B, Chen M, Wang J, Tian Z, He Y, Lin D. 2011. "A brief utilization report on the Illumina HiSeq 2000 sequencer." Mycology 2: 169-191.
-
7. Lu R, Lin Z, Zhang Y, Chai X, Dong Y, Xiao G, Zhang J-M, Fang J, Hu L-H. 2008. "Comparison on the karyotypes of Scapharca subcrenata, Tegillarca granosa and Estellarca olivacea." Journal of Shanghai Fisheries University 17: 625-629.
-
8. Manee MM, Al-Shomrani BM, Al-Fageeh MB. 2020. "Genome-wide characterization of simple sequence repeats in Palmae genomes." Genes Genomics 42: 597-608.
-
9. Marcais G, Kingsford C. 2011. "A fast, lock-free approach for efficient parallel counting of occurrences of k-mers." Bio- informatics 27: 764-770.
-
10. Meglecz E, Pech N, Gilles A, Dubut V, Hingamp P, Trilles A, Grenier R, Martin JF. 2014. "QDD version 3.1: a user-friendly com- puter program for microsatellite selection and primer design revisited: experimental validation of variables determining genotyping success rate." Mol Ecol Resour 14: 1302-1313.
-
11. Mohamat-Yusuff F, Zulkarnain Z, Anuar NZA, Joni AAM, Kusin FM, Mohamed KN, Zulkeflee Z, Asha'ari ZH, Zulkifli SZ, Arshad A, Ismail A. 2020. "Impact of Diuron contamination on blood cockles (Tegillarca granosa Linnaeus, 1758)." Mar Pollut Bull 161: 111698.
-
12. Penaloza C, Bishop SC, Toro J, Houston RD. 2014. RAD Sequencing reveals genome-wide heterozygote deficiency in pair crosses of the Chilean mussel Mytilus spp. Proceedings of the World Congress on Genetics Applied to Livestock Production. Species Breeding: Breeding in Aquaculture Species: 275.
-
13. Rozen S, Skaletsky H. 2000. "Primer3 on the WWW for general users and for biologist programmers." Methods Mol Biol 132: 365-386.
-
14. Su W, Zha S, Wang Y, Shi W, Xiao G, Chai X, Wu H, Liu G. 2017. "Benzo[a]pyrene exposure under future ocean acidification scenarios weakens the immune responses of blood clam, Tegillarca granosa." Fish Shellfish Immunol 63: 465-470.
-
15. Vurture GW, Sedlazeck FJ, Nattestad M, Underwood CJ, Fang H, Gurtowski J, Schatz MC. 2017. GenomeScope: fast reference-free genome profiling from short reads. Bioinformatics 33: 2202-2204.
-
16. Wang Y, Zeng Q, Xu L. 2013. "Population structure of the blood clam (Tegillarca granosa) in China based on microsatellite markers." Genet Mol Res 12: 892-900.
-
17. Zimin AV, Marçais G, Puiu D, Roberts M, Salzberg SL, Yorke JA. 2013. "The MaSuRCA genome assembler." Bioinformatics 29: 2669-2677.
Copyright (C) 2025. The Korean Society of Marine Life Science